Protaktyn
| tor ← protaktyn → uran | |||||||||||||||||||||||||||||||||||||
| Wygląd | |||||||||||||||||||||||||||||||||||||
| srebrzystobiały | |||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
Widmo emisyjne protaktynu | |||||||||||||||||||||||||||||||||||||
| Ogólne informacje | |||||||||||||||||||||||||||||||||||||
| Nazwa, symbol, l.a. |
protaktyn, Pa, 91 | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grupa, okres, blok | |||||||||||||||||||||||||||||||||||||
| Stopień utlenienia |
II, III, IV, V | ||||||||||||||||||||||||||||||||||||
| Właściwości metaliczne | |||||||||||||||||||||||||||||||||||||
| Właściwości tlenków | |||||||||||||||||||||||||||||||||||||
| Masa atomowa | |||||||||||||||||||||||||||||||||||||
| Stan skupienia |
stały | ||||||||||||||||||||||||||||||||||||
| Gęstość |
15,37 g/cm³[3] | ||||||||||||||||||||||||||||||||||||
| Temperatura topnienia |
1567 °C[1] | ||||||||||||||||||||||||||||||||||||
| Temperatura wrzenia |
4227 °C[1] | ||||||||||||||||||||||||||||||||||||
| Numer CAS | |||||||||||||||||||||||||||||||||||||
| PubChem | |||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
| Jeżeli nie podano inaczej, dane dotyczą warunków normalnych (0 °C, 1013,25 hPa) | |||||||||||||||||||||||||||||||||||||

Protaktyn (Pa, łac. protactinium) – pierwiastek chemiczny z grupy aktynowców o liczbie atomowej 91. Jest ciężkim srebrnoszarym metalem, łatwo reagującym z tlenem, parą wodną i kwasami nieorganicznymi. Tworzy związki chemiczne, w których jest zazwyczaj na V stopniu utlenienia, ale może także przyjmować II, III i IV stopień. Średnie stężenie protaktynu w skorupie ziemskiej jest zazwyczaj rzędu kilku ppt, ale może osiągać stężenia do kilku ppm w pewnych złożach rud uraninitowych. Ze względu na rzadkość, wysoką radioaktywność i toksyczność, metal ten obecnie nie posiada zastosowania poza badaniami naukowymi, do których pozyskiwany jest ze zużytego paliwa jądrowego.
Protaktyn po raz pierwszy został zidentyfikowany w 1913 roku przez Kazimierza Fajansa i Oswalda Helmutha Göhringa, którzy nazwali go brevium ze względu na krótki czas połowicznego rozpadu badanego izotopu 234Pa. Bardziej stabilny izotop protaktynu został odkryty w 1918 roku, a jego nazwę zmieniono na protoaktyn, a następnie, w 1949 roku, na „protaktyn”, co oznacza „rodzic aktynu” i odnosi się do faktu, że produktem rozpadu protaktynu jest aktyn.
Najtrwalszym i najbardziej rozpowszechnionym (niemal 100%) naturalnie występującym izotopem protaktynu jest 231Pa, będący produktem rozpadu 235U, ma okres półtrwania ok. 32 530 lat[4]. Znacznie mniejsze, śladowe ilości krótkotrwałego 234mPa występują w produktach szeregu promieniotwórczego 238U. 233Pa jest produktem rozpadu 233Th, będącego częścią łańcucha przemian wykorzystywanych do otrzymania 233U poprzez napromieniowanie neutronami 232Th. Jest niepożądanym produktem pośrednim w opartych na torze reaktorach jądrowych i jest usuwany z aktywnego obszaru reaktora podczas procesu powielania.
Analiza względnych stężeń różnych izotopów uranu, toru i protaktynu w wodzie i minerałach jest używana w datowaniu izotopowym osadów, których wiek sięga do 175 tys. lat oraz w modelowaniu różnych procesów geologicznych.
Występowanie
[edytuj | edytuj kod]Protaktyn występuje w blendzie smolistej (uraninicie) w stężeniu w rudzie około 0,3–3 ppm. Jest jednorodnie rozprzestrzeniony w większości substancji naturalnych, w tym w wodzie, gdzie występuje w mniejszych stężeniach, rzędu 1 ppt, co odpowiada promieniotwórczości 0,1 pCi/g. W glebie piaszczystej występuje około 500-krotnie więcej protaktynu niż w wodzie, nawet jeśli woda ta jest obecna w tej glebie. Znacznie wyższe krotności, rzędu 2000 i powyżej, zostały zmierzone w piaskach gliniastych i glinach, takich jak bentonit. Protaktyn jest jednym z najrzadszych i najdroższych pierwiastków występujących w przyrodzie.
Źródła naturalne
[edytuj | edytuj kod]
Ponieważ czas połowicznego rozpadu 231Pa jest krótki w porównaniu do skali geologicznej, jego występowanie jest ściśle związane z występowaniem 235U. Zawartość uranu w skorupie ziemskiej jest na poziomie 2,7 ppm[5], z czego 0,711% to 235U, stąd naturalne rozpowszechnienie 231Pa (obliczone z okresów połowicznego rozpadu 231Pa i 235U) wynosi 0,87×10−6, tylko nieznacznie mniej niż w przypadku 226Ra. Przyjmując, że masa skorupy ziemskiej (do głębokości 36 km) wynosi 2,5×1025 g[6], to całkowite zasoby protaktynu wynoszą 2,2×107 ton.
Wyraźna tendencja protaktynu do hydrolizy jest podstawą metody datowania osadów morskich do ich wieku sięgającego miliona lat[7]. Przy niezakłóconym uformowaniu geologicznym stosunek Pa:U wynosi 3,2×10−7, ale współczynnik ten jest zakłócany, kiedy złoże jest wymywane przez wody gruntowe, które przenoszą uran do morza. Przy pH wody morskiej protaktyn i tor hydrolizują i odkładają się na dnie oceanicznym, pozostawiając uran w roztworze jako U2+2. Jako że rozpad 231Pa i 230Th zachodzi z różnymi prędkościami, to wzajemny współczynnik zawartości tych pierwiastków na różnych głębokościach może być użyty do określenia szybkości sedymentacji[7].
Do określenia zawartości 231Pa w ciałach stałych używana jest nowa metoda, o rząd wielkości dokładniejsza niż metody oparte na zliczaniu rozpadów, która oparta jest na spektroskopii masowej jonizacji termicznej (TIMS) protaktynu w węglanach. Dzięki tej metodzie mogą być datowane węglany o wieku 10–250 tys. lat[7].
233Pa nie został wykryty w naturze, ale ślady 237Np, jego substratu, jak i 225Ac, produktu jego rozpadu, zostały wykryte w ściekach rafinerii uranu[8]. Można wnioskować, że 233Pa jest ciągle naturalnie tworzony w reakcji:
- 238U(n,2n) → 237U(β−;6,75 dnia) → 237Np(α;2,14×106 lat) → 233Pa
W reaktorach atomowych
[edytuj | edytuj kod]Dwa ważne izotopy protaktynu, 231Pa i 233Pa powstają z toru w reaktorach jądrowych; oba są niepożądane i są zazwyczaj usuwane, tym samym komplikując schemat i działanie reaktora jądrowego. W szczególności 232Th poprzez reakcje n,2n tworzy 231Th, który szybko (t1/2 = 25,5 godz.) rozpada się do 231Pa. Izotop ten ma stosunkowo długi okres połowicznego zaniku, równy 32 760 lat, i jest główną przyczyną długotrwałej radiotoksyczności zużytego paliwa jądrowego. Protaktyn-233 jest tworzony z 232Th poprzez wychwyt neutronu. Następnie rozpada się do uranu-233 lub wychwytuje następny neutron i przekształca się w bardzo trwały uran-234. 233Pa ma względnie długi okres połowicznego zaniku równy 27 dni oraz wysoki przekrój czynny dla wychwytu neutronów (tak zwana „trucizna reaktorowa”). Stąd też, zamiast szybko rozpadać się do użytecznego 233U, znacząca część 233Pa przekształca się w słabo rozszczepialne izotopy i pochłania neutrony, zmniejszając tym samym wydajność reaktora. By temu zapobiec, 233Pa jest usuwany z aktywnej strefy pracy torowych reaktorów chłodzonych stopionymi solami, by tor ulegał rozpadowi do 233U. Jest to osiągane poprzez użycie kilkumetrowej wysokości kolumn stopionego bizmutu z rozpuszczonym w nim licie. W uproszczonym schemacie można to przedstawić, że lit wybiórczo redukuje sole protaktynu do metalicznego protaktynu, który następnie jest wyodrębniany ze stopionych soli, a bizmut służy tylko jako rozpuszczalnik. Wybierany jest ze względu na niską temperaturę topnienia (271 °C), niską prężność pary, dobrą rozpuszczalność litu i aktynowców oraz niemieszalność z roztopionymi chlorkami.
Izotopy protaktynu
[edytuj | edytuj kod]Obecnie (sierpień 2012) znanych jest 29 izotopów protaktynu, spośród których tylko trzy mają znaczenie naukowe;231Pa, 233Pa i 234Pa. Dwa z nich występują naturalnie – 231Pa (~100%) i w śladowych ilościach 234Pa, który istnieje ponadto w dwóch różnych stanach energetycznych. 233Pa jest produktem pracy reaktorów atomowych[4]. Protaktyn jest źródłem cząstek alfa. Niemal cały (99,8%) uran-238 rozpada się najpierw do izomeru 234mPa.
Protaktyn-231
[edytuj | edytuj kod]231Pa jest źródłem promieniowania α, a jego masa atomowa została ustalona na 231,03588 ±0,00002[9]. W szeregu uranowo-aktynowym jest produktem rozpadu 231Th, a dalej rozpada się na 227Ac, od którego zaczerpnął swoją nazwę. Potwierdzone okresy półtrwania wynoszą od 32 000±10%[10] do 34 300 ±300 lat. Ostatnie badania ustaliły okres półtrwania na ok. 35 530 ±250 lat (z 95% pewnością)[11]. Dlatego też 231Pa jest jedynym izotopem dostępnym w miligramowych ilościach. Przekrój czynny wychwytu neutronów termicznych wynosi 211 ±2 barnów. Okres półtrwania samorzutnego rozszczepienia wynosi 1,1×1016 lat.[11]
231Pa może być wyizolowany z przetworzonych rud uranowych lub wytworzony na drodze dwóch reakcji jądrowych: 232Th(n,2n)231Th lub 230Th(n,γ)231Th. W zasadzie powinno to wyeliminować wszystkie problemy związane z otrzymywaniem 231Pa. Jednakże napromieniowanie neutronami 232Th daje duże ilości 233Pa i innych niepożądanych zanieczyszczeń, a względnie małe ilości 231Pa[11].
Protaktyn-233
[edytuj | edytuj kod]233Pa jest jedynym izotopem protaktynu, który otrzymywany jest w gramowych ilościach; pierwszy gram został otrzymany w 1964 roku przez naukowców w National Reactor Testing Station w Idaho[4]. Znaczenie tego izotopu wynika z faktu, że jest etapem pośrednim w produkcji rozszczepialnego 233U. Reakcja została odkryta w 1938 roku przez Lise Meitner[12]:
- 232Th(n,γ)233Th(β−,22 min)→233Pa(β−, 27 dni) →233U
233Pa wyparł izotop 234Pa z zastosowań jako znacznik izotopowy ze względu na swój okres półtrwania, stosunkową łatwość otrzymywania oraz nietrudne do wykrycia widmo gamma[13][14].
Protaktyn-234
[edytuj | edytuj kod]Nuklid 234Pa występuje naturalnie w dwóch postaciach izomerycznych: 234mPa, odkryty przez Fajansa i Göhringa w 1913 roku oraz 234Pa, odkryty przez Hahna w 1921 roku. Oba izotopy są źródłami promieniowania β−, rozpadając się do 234U, ale 234mPa jest metastabilny i w 0,13% rozpadów, w wyniku przejścia izomerycznego, rozpada się do swojego stanu podstawowego, 234Pa[11].
Otrzymywanie i oczyszczanie
[edytuj | edytuj kod]Ze względu na niewielkie zapotrzebowanie na protaktyn, nigdy nie uzyskiwano 231Pa z rud rodzimych na dużą skalę. Mierzalne ilości protaktynu zawsze były otrzymywane z pozostałości oczyszczania uranu. Ze względów ekonomicznych nie zawsze jest możliwa optymalizacja wydzielania protaktynu z wszystkich odpadów jądrowych i pozostałościach z innych procesów. Zazwyczaj protaktyn jest frakcjonowany na każdym z etapów wzbogacania i wydobywania uranu z jego rud.
Przed rozwojem energii atomowej, blenda smolista była przetwarzana ze względu na zawartość radu. Rozdrobniona ruda, po wcześniejszym wyprażeniu z Na2CO3, była ługowana wodnymi roztworami H2SO4 lub HNO3 (lub ich mieszaniną), a substancje nierozpuszczalne w kwasach były roztwarzane przy użyciu roztworów NaOH lub Na2CO3. Pozostałość była następnie ługowana kwasem solnym, aby odzyskać rad[7]. Pozostałość zawierała większą lub mniejszą część pierwotnego protaktynu, w zależności od względnych proporcji kwasów używanych w roztwarzaniu; wyższe stężenie H2SO4 i ogólna całkowita kwasowość sprzyja przechodzeniu protaktynu do roztworu. Metoda ta (Rückrückstände) była użyta przy odkryciu 231Pa przez Hahna i Meitner (1918), a później do otrzymania pierwszych miligramowych ilości pierwiastka (Grosse, 1927) oraz do otrzymania 0,5 g tego pierwiastka przez Grauego i Kädinga w 1934 roku[7].
Po II wojnie światowej do oczyszczania uranu używany był proces ekstrakcji eterowej. Kwasowy roztwór powstały po roztwarzaniu rudy był traktowany Na2CO3, aby strącić mniej zasadowe metale, zostawiając przy tym uran w roztworze jako kompleks węglanowy. Katzin oszacował, że strącony osad węglanowy zawiera 0,30–0,35 ppm protaktynu[15], a dalsze przetworzenie tego osadu dało ok. 25 mg czystego metalu. Kiedy proces został zmodyfikowany by wyeliminować strącanie osadu węglanowego, protaktyn przechodził przez etap ekstrakcji eterowej do wodnego rafinatu, co pozwoliło na odzyskanie 35 mg czystej substancji. Dalsze modyfikacje procesu pozwoliły na strącanie w sposób ciągły osadu, który zawierał niemal cały protaktyn. Osad ten, był okresowo odfiltrowywany, co przyniosło ok. 2 g protaktynu[7].
Wodny rafinat z ekstrakcji eterowej był poddawany działaniu wapna, a odfiltrowany osad był magazynowany do przyszłego odzysku uranu i innych rynkowo cennych metali. Zgromadzony materiał był następnie poddawany kilkuetapowemu procesowi: roztwarzanie za pomocą kwasu siarkowego, następnie ekstrakcja z użyciem HDEHP, a ostatecznie reekstrakcja roztworem węglanu sodu. Roztwory ściekowe i osady były zrzucane do basenów ściekowych, gdzie tracono znaczną część protaktynu i 230Th. Proces przeszedł w 1972 roku modyfikację poprzez dodanie odpowiedniej ilości wodorotlenku sodu do roztworu Na2CO3, powodując całkowite strącenie uranu, tym samym minimalizując dalsze straty Pa i 230Th. Odfiltrowany osad (koncentrat Cottera) składał się z 2000 ton (suchej masy) mieszaniny tlenków i węglanów i zawierał około 30 ton U3O8, 14 kg 230Th i 75 g protaktynu. Materiał ten został przetworzony w Laboratoriach Mound[7].
W zakładach Windscale należącej do UKAEA, po usunięciu siarczanowego osadu zawierającego rad, roztwór był buforowany do pH ok. 2, a uran był strącany poprzez dodawanie nadtlenku wodoru. Osad nadtlenkowy niósł z sobą ponad 80% protaktynu, który był następnie ponownie rozpuszczany w kwasie azotowym. Niskie pH sprzyjało tworzeniu zawierającego protaktyn krzemionkowego szlamu, który osadzał się na ściankach instalacji ekstrakcji eterowej. Ten eterowy szlam był gromadzony do dalszego odzyskiwania uranu. Metoda ta była najbogatszym źródłem protaktynu, ostatecznie dając 127 g czystego pierwiastka[16].
| Rückrückstände[17] | Szlam eterowy[18] | Koncentrat Cottera[7] | |||
|---|---|---|---|---|---|
| Składnik | Zawartość (%) | Składnik | Zawartość (%) | Składnik | Zawartość (%) |
| SiO2 | 60 | U | 28,3 | U3O8 | 13,8 |
| Fe2O3 | 22 | Fe | 7,7 | Fe | ok. 30 |
| PbO | 8 | Si | 6,4 | Si | ok. 4 |
| Al2O3 | 5 | Ba | ok. 3 | Na | ok. 60 |
| MnO | 1 | Zr | 2,7 | Mo | ≤2 |
| CaO | 0,6 | Mo | 2,7 | V | ≤1 |
| MgO | 0,5 | F− | 1,8 | Al | ≥0,3 |
| Ti | 0,3 | NH+4 | 1,7 | Th | 0,15 |
| Zr | 0,1 | Ca | 1,5 | Ti | 0,1 |
| HF i inne | – | V | 0,9 | Ca | 0,07 |
| grafit | 0,1 | Ti | 0,44 | Cu | 0,05 |
| Pa2O5 | <3×10−3 | Pb | 0,4 | Zr | 0,04 |
| Al | 0,27 | Mg | 0,04 | ||
| P | 0,15 | Ni | 0,03 | ||
| Sr | 0,09 | Mn | 0,01 | ||
| Nb, Ta | <0,1 | Cr | 0,01 | ||
| Mg, Ni, Cr | <0,01 | B | 0,002 | ||
| Mn, Co, Mn, Sn | <0,01 | Be | 7×10−4 | ||
| Pa | 3,7×10−4 | Pa | 4×10−5 | ||
Obecnie w większości produkowany jest jako produkt pośredni reakcji jądrowych w wysokotemperaturowych reaktorach torowych podczas otrzymywania 233U:
Wydzielenie czystego protaktynu
[edytuj | edytuj kod]Protaktyn został wyizolowany przez Aristida van Grossego (1893–1976), początkowo w postaci tlenku Pa2O5 (2 mg) w 1927 roku, a w roku 1934 jako wolny pierwiastek, z 0,15 mg Pa2O5. Van Grosse określił masę atomową pierwiastka na 230,6 ±0,5[4]. Zastosował dwie różne metody: w pierwszej, tlenek protaktynu był napromieniowywany w próżni wiązką elektronów o energii 35 keV. W drugiej metodzie, nazywaną procesem van Arkela-de Boera, tlenek został chemicznie przekształcony do halogenku (chlorku, bromku lub jodku), a następnie rozłożony termicznie w próżni z użyciem elektrycznie rozgrzanego włókna:
- 2PaI5 → 2Pa + 5I2
W tym samym roku Graue i Käding otrzymali 0,5 g protaktynu w postaci K2PaF7 z 5,5 tony resztek blendy smolistej[19].
Rozwój energii atomowej doprowadził do przetworzenia dużych ilości rud o wysokiej zawartości uranu oraz spowodował nagromadzenie odpadów promieniotwórczych. W 1961 roku w zakładzie paliw jądrowych Springfields, należącym do United Kingdom Atomic Energy Authority (UKAEA, Komisja Nadzoru Badań i Wykorzystania Energii Atomowej Zjednoczonego Królestwa), wyizolowano 127 g protaktynu o czystości 99,9% poprzez 12-etapowy proces przetworzenia 60 ton odpadów promieniotwórczych o zawartości protaktynu na poziomie 4 ppm[4]. Ze względu na to, że odpady zawierały też 12 ton uranu, było ekonomicznie uzasadnione odzyskanie obu pierwiastków, większość kosztów przypadło na odzysk uranu[4]. Całkowity koszt procesu wyniósł około 500 tys. dolarów. Przez wiele lat, ze względu na oferowanie przez UKAEA protaktynu w symbolicznych cenach, było to jedyne znaczące źródło tego metalu, który był wykorzystywany w różnych laboratoriach do badań naukowych[4].
Właściwości
[edytuj | edytuj kod]Właściwości fizyczne
[edytuj | edytuj kod]Protaktyn należy do grupy aktynowców i jest położony w układzie okresowym na lewo od uranu i na prawo od toru, co powoduje, że wiele jego właściwości fizycznych jest pośrednich pomiędzy tymi dwoma pierwiastkami. Stąd też protaktyn ma większą gęstość i twardość niż tor, ale jest lżejszy niż uran, a jego temperatura topnienia jest niższa niż toru, a wyższa niż uranu. Rozszerzalność cieplna, przewodność cieplna i elektryczna tych trzech pierwiastków jest porównywalna do wartości tych wielkości dla metali nieszlachetnych. Przybliżona wartość modułu Kirchhoffa protaktynu jest podobna do tytanu. Przy schładzaniu w temperaturze pokojowej protaktyn krystalizuje do przestrzennie centrowanej struktury tetragonalnej[20], która może być uważana za zniekształconą przestrzennie centrowaną regularną sieć krystaliczną; sieć ta nie zmienia się do ciśnienia 53 GPa. Struktura zmienia się w regularną ściennie centrowaną (fcc) podczas schładzania z wysokiej temperatury przy ok. 1200 °C. Teoretyczne wyliczenia przewidują, że metaliczny protaktyn podlega przejściu fazowemu do rombowej struktury α-uranu poniżej 100 GPa. Przy bardzo wysokich ciśnieniach metal osiąga ściśle upakowaną strukturę hcp. Współczynnik rozszerzalności cieplnej fazy tetragonalnej pomiędzy temperaturą pokojową a 700 °C wynosi 9,9×10−6/°C.
Protaktyn jest paramagnetykiem, a w temperaturze poniżej 1,4 K staje się nadprzewodnikiem. Tetrachlorek protaktynu (PaCl4), jest paramagnetykiem w temperaturze pokojowej, ale przy schłodzeniu do 182 K staje się ferromagnetykiem.
Właściwości chemiczne
[edytuj | edytuj kod]Protaktyn jest metalem o srebrnoszarym połysku, który utrzymuje się przez kilka miesięcy w atmosferze powietrza[21]. Niewielka utrata metalicznego połysku została zaobserwowana po ogrzewaniu przez godzinę próbki protaktynu do temperatury 100 °C. Ogrzewanie próbki przez godzinę do temperatury 300 °C powoduje zmianę barwy na szarobiałą i rozpoczyna rozpad próbki. Metal przy 300 °C wystawiony na działanie tlenu, pary wodnej i dwutlenku węgla jako produkt daje Pa2O5; reakcje z NH3 i H2 dały odpowiednio PaN2 i PaH3[22]. Pierwiastek występuje głównie w dwóch stopniach utlenienia, IV i V, oba w ciałach stałych i roztworach, a stopnie II i III w pewnych fazach stałych. Jako że konfiguracja elektronowa atomu protaktynu to [Rn]7s26d15f2, V stopień utlenienia odpowiada niskoenergetycznej (dlatego też uprzywilejowanej) konfiguracji radonu. Związki Pa(IV) i Pa(V) łatwo tworzą w wodzie wodorotlenki i hydroksosole, wśród których dominują Pa(OH)3+, Pa(OH)2+2, Pa(OH)+3 i Pa(OH)4. Roztwory zawierające te jony są bezbarwne. Innymi znanymi jonami protaktynu są m.in.PaCl2+2, PaSO2+4, PaF3+, PaF2+2, PaF6−, PaF2−7 i PaF3−8.
Protaktyn może tworzyć stopy. Otrzymano stopy protaktynu z metalami szlachetnymi poprzez redukcję Pa2O5 za pomocą wodoru w obecności platyny, irydu i rodu[23]. Możliwe jest także otrzymanie stopu protaktynu z berylem, Be13Pa.
Proste związki protaktynu
[edytuj | edytuj kod]Znane są trzy tlenki protaktynu: PaO, PaO2 i Pa2O5.
Biały Pa2O5 powstaje, gdy uwodniony tlenek Pa2O5·nH2O, jak i wiele innych związków protaktynu, jest ogrzewany w atmosferze powietrza lub tlenu do temperatury powyżej 500–650 °C[22]. Czarny PaO2 jest otrzymywany w reakcji redukcji Pa2O5 wodorem w temperaturze 1550 °C. PaO tworzy się na powierzchni metalicznego protaktynu[22].
Tlenki potrójne (składające się z trzech pierwiastków) zostały otrzymane w reakcji PaO2 i Pa2O5 z tlenkami innych pierwiastków[24][25].
Podczas dodawania H2O2 do roztworu Pa(V) w 0,25 M H2SO4 strąca się jasnożółty osad Pa2O9·3H2O, będący niestabilnym nadtlenkiem protaktynu. Skład nadtlenku opisanego wzorem Pa2Ox·3H2O zmienia się stopniowo wraz z czasem od x=9 do x=5[26].
W reakcji wodoru z metalicznym protaktynem w temperaturze 250 °C i ciśnieniu 600 torów otrzymano w 1954 roku[22] czarną substancję izostrukturalną z β-UH3. Związek tan ma strukturę regularną o stałych sieciowych a = (6,648 ±0,005) Å. W 1972 roku otrzymano[21] w temperaturze 100, 200 i 300 °C szarą substancję w postaci proszku, izostrukturalną z α-UH3. Stała sieciowa dla α-PaH3 otrzymanego przy 100 i 200 °C wynosi a = (4,150 ±0,002) Å, natomiast dla produktu otrzymanego przy 300 °C a = (4,154 ±0,002) Å.
Węglik protaktynu(IV), PaC, uzyskano[27] poprzez redukcję Pa2O5 z grafitem przy zmniejszonym ciśnieniu w temperaturze powyżej 1200 °C. Produkt otrzymany przy 1950 °C miał strukturę fcc (typ NaCl) o stałej a = (5,0608 ±0,0002) Å. Przy 2200 °C zaobserwowano słabe linie odpowiadające PaC2, a tetragonalna struktura otrzymanego związku charakteryzowała się stałymi sieciowymi a = (3,61 ±0,01) Å i c = (6,11 ±0,01) Å.
Halogenki i tlenohalogenki protaktynu(V) i protaktynu(IV) są otrzymywane z wykorzystywaniem jako substratu wodnego roztworu Pa(V). Do uzyskania halogenków metalu na V i IV stopniu utlenienia wykorzystywany jest PaC. Z halogenków protaktynu(III) znany jest tylko PaI3, otrzymany poprzez ogrzewanie PaI5 przez kilkanaście dni w temperaturze 360–380 °C przy ciśnieniu 10−6 tora.
Wszystkie podwójne halogenki są lotne przy umiarkowanych temperaturach, co było wykorzystywane przy rozdzielaniu 233Pa od ThO2 i otrzymywaniu czystego 231Pa i 234Pa[28].
Historia
[edytuj | edytuj kod]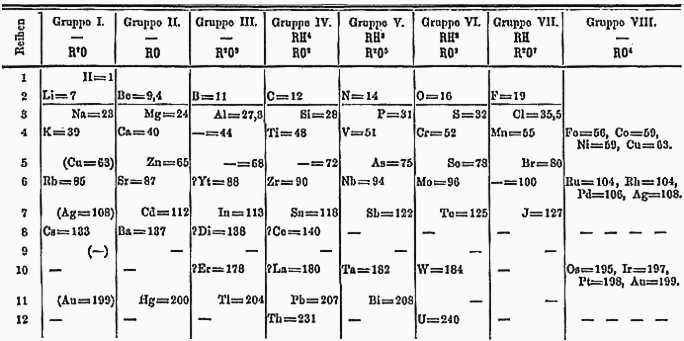
W 1871 roku Dmitrij Mendelejew przewidział istnienie ośmiu pierwiastków pomiędzy torem a uranem. W tamtym okresie grupa aktynowców była nieznana, stąd też uran był położony poniżej wolframu, a tor poniżej cyrkonu, pozostawiając puste miejsce poniżej tantalu. Układ okresowy pierwiastków był przedstawiany w takiej postaci aż do lat 50. XX wieku. Przez długi czas chemicy poszukiwali eka-tantalu – pierwiastka mającego właściwości zbliżone do tantalu, co uczyniło odkrycie protaktynu niemal niemożliwym.
W 1900 roku William Crookes wydzielił z uranu protaktyn jako silnie promieniotwórczą substancję, jednak nie był w stanie opisać jej jako nowy pierwiastek chemiczny, stąd też nazwał ją uranem-X[29]. Crookes rozpuścił azotan uranylu w eterze, otrzymując tym samym w fazie wodnej głównie jony 23490Th i 23491Pa. Jego metoda była używana do otrzymywania 23490Th i 23491Pa ze związków uranu aż do lat 50. XX wieku[30]. Protaktyn po raz pierwszy został zidentyfikowany w 1913 roku, kiedy Kazimierz Fajans i Oswald Göhring natrafili na izotop 234Pa podczas swoich badań nad szeregiem promieniotwórczym uranu-238:
- 238U → 234Th → 234Pa → 234U
Nowy pierwiastek został nazwany brevium (od łacińskiego słowa brevis oznaczającego chwilę, krótki okres), z powodu jego krótkiego okresu połowicznego zaniku[31][32]. W 1918 roku dwie grupy naukowców, kierowane przez Otto Hahna i Lise Meitner w Niemczech oraz Fredericka Soddy’ego i Johna Cranstona w Wielkiej Brytanii, niezależnie od siebie odkryły inny izotop protaktynu, 231Pa, charakteryzujący się okresem półtrwania wynoszącym ok. 32 tys. lat. Po tym odkryciu nazwa pierwiastka została zmieniona z „brevium” na „protoactinium”, z uwagi na występowanie jego w szeregu uranu-235 przed aktynem. Ze względu na łatwość wymowy, nazwa została zmieniona w 1949 roku przez IUPAC na protactinium[4]. Odkrycie protaktynu zapełniło ostatnią lukę we wczesnej wersji układu okresowego zaproponowanego przez Mendelejewa w 1869 roku oraz przyniosło sławę naukowcom zaangażowanym w jego odkrycie, szczególnie Meitner[33].
Zastosowanie protaktynu
[edytuj | edytuj kod]Protaktyn jest używany do produkcji scyntylatorów do wykrywania promieniowania rentgenowskiego, składających się z tlenków gadolinu, protaktynu, cezu, metali ziem rzadkich i innych pierwiastków. Scyntylatory te są używane szczególnie w aparaturze do tomografii komputerowej[34].
Tlenki mieszane Nb, Mg, Ga i Mn, domieszkowane 0,005–0,52% Pa2O5 są używane jako wysokotemperaturowe dielektryki (do 1300 °C) w kondensatorach ceramicznych[35].
Stosunek zawartości 231Pa/235U jest elementem wykorzystywanym do określania wieku próbek w datowaniu metodą uranowo-torową.
Toksyczność
[edytuj | edytuj kod]231Pa jest niebezpieczny dla organizmów, podobnie jak inne źródła promieniowania α o podobnym okresie połowicznego zaniku. W organizmie gromadzi się w nerkach i kościach. Maksymalna ilość protaktynu, która nie jest traktowana jako niebezpieczna po absorpcji przez organizm wynosi 0,03 μCi, co odpowiada masie 0,5 μg 231Pa. Protaktyn znajdujący się w powietrzu jako aerozol jest 250 mln razy bardziej toksyczny niż cyjanowodór[36]. Z tego powodu wszystkie operacje z mierzalnymi ilościami protaktynu powinny być przeprowadzane w specjalnych izolowanych komorach.
Uwagi
[edytuj | edytuj kod]- ↑ Podana wartość stanowi przybliżoną standardową względną masę atomową (ang. abridged standard atomic weight) publikowaną wraz ze standardową względną masą atomową, która wynosi 231,03588 ± 0,00001. Zob. Prohaska i in. 2021 ↓, s. 584.
Przypisy
[edytuj | edytuj kod]- ↑ a b c Per. Enghag: Encyclopedia of the elements. Technical data, history, processing, application. Weinheim: Wiley-VCH, 2004, s. 1163–1165. ISBN 3-527-30666-8.
- ↑ Thomas Prohaska i inni, Standard atomic weights of the elements 2021 (IUPAC Technical Report), „Pure and Applied Chemistry”, 94 (5), 2021, s. 573–600, DOI: 10.1515/pac-2019-0603 (ang.).
- ↑ C.R. Hammond, The elements. Protactinium, [w:] David R. Lide (red.), CRC Handbook of Chemistry and Physics, wyd. 90, Boca Raton: CRC Press, 2009, s. 4-28–4-29, ISBN 978-1-4200-9084-0 (ang.). Wartość obliczona.
- ↑ a b c d e f g h Protactinium. W: The Chemistry of the Actinide and Transactinide Elements. Dordrecht: Springer Netherlands, 2011, s. 163–164. ISBN 978-94-007-0211-0. (ang.).
- ↑ S.R. Taylor, Abundance of chemical elements in the continental crust: a new table, „Geochimica et Cosmochimica Acta”, 28 (8), 1964, s. 1273–1285, DOI: 10.1016/0016-7037(64)90129-2 [dostęp 2022-06-30] (ang.).
- ↑ Karl Hans Wedepohl: Handbook of geochemistry. T. 1. 1969. ISBN 3-540-06578-4. (ang.).
- ↑ a b c d e f g h i Protactinium. W: The Chemistry of the Actinide and Transactinide Elements. Dordrecht: Springer Netherlands, 2011, s. 170–175. ISBN 978-94-007-0211-0. (ang.).
- ↑ D.F. Peppard i inni, Occurrence of the (4n + 1) Series in Nature, „Journal of the American Chemical Society”, 74 (23), 1952, s. 6081–6084, DOI: 10.1021/ja01143a074 [dostęp 2022-06-30] (ang.).
- ↑ J.R. De Laeter, K.G. Heumann, K.J.R. Rosman, Isotopic Compositions of the Elements 1989, „Journal of Physical and Chemical Reference Data”, 20 (6), 1991, s. 1327–1337, DOI: 10.1063/1.555903 [dostęp 2022-06-30] (ang.).
- ↑ Aristid v. Grosse, Über die Halbwertszeit des Protactiniums, „Naturwissenschaften”, 20 (27), 1932, s. 505–505, DOI: 10.1007/BF01505061 [dostęp 2022-06-30] (niem.).
- ↑ a b c d Protactinium. W: The Chemistry of the Actinide and Transactinide Elements. Dordrecht: Springer Netherlands, 2011, s. 166–170. ISBN 978-94-007-0211-0. (ang.).
- ↑ Ruth Lewin Sime: Lise Meitner:A Life in Physics. California Studies in the History of Science, czerwiec 1997, s. 456. ISBN 978-0-520-20860-5. (ang.).
- ↑ C.E. Crouthamel, F. Adams, R. Dams: Applied gamma-ray spectrometry. Oxford, New York, Pergamon Press, 1970. ISBN 0-08-006888-X. (ang.).
- ↑ C.W. Sill, Preparation of Protactinium-233 Tracer., „Analytical Chemistry”, 38 (11), 1966, s. 1458–1463, DOI: 10.1021/ac60243a004 [dostęp 2022-06-30] (ang.).
- ↑ L.I. Katzin, Q. Van Winkle, J. Sedlet, Analysis of Ore Residues for Ionium and Protactinium 1, „Journal of the American Chemical Society”, 72 (10), 1950, s. 4815–4817, DOI: 10.1021/ja01166a512 [dostęp 2022-06-30] (ang.).
- ↑ A.G. Goble, A.G. Maddock, Protactinium—III solvent extraction from halide solutions, „Journal of Inorganic and Nuclear Chemistry”, 7 (1-2), 1958, s. 94–112, DOI: 10.1016/0022-1902(58)80032-9 [dostęp 2022-06-30] (ang.).
- ↑ A.V. Grosse, M Agruss. „Industrial & Engineering Chemistry Research”. 27, s. 422–426, 1935. (ang.).
- ↑ J.S. Nairn, D.A. Collins. „Proceedings of the Second United Nations International Conference on the Peaceful uses of Atomic Energy”. 17, s. 216–235, 1–13 września 1958. Genewa. (ang.).
- ↑ Georg Graue, Hans Käding, Reinherstellung von einem halben Gramm Protaktinium (Element 91), „Die Naturwissenschaften”, 22 (22-24), 1934, s. 386–388, DOI: 10.1007/BF01503626 [dostęp 2022-06-30] (niem.).
- ↑ J. Donohue, On the crystal structure of protactinium metal, „Acta Crystallographica”, 12 (9), 1959, s. 697–698, DOI: 10.1107/S0365110X59002031 [dostęp 2022-06-30] (ang.).
- ↑ a b Raymond Lloyd Dod. Some Properties of Protactinium Metal and Its Compounds. „US report LBL-659”, maj 1972. Lawrence Berkeley Laboratory: University of California. (ang.).
- ↑ a b c d Philip A. Sellers i inni, The Preparation of Some Protactinium Compounds and the Metal 1, „Journal of the American Chemical Society”, 76 (23), 1954, s. 5935–5938, DOI: 10.1021/ja01652a011 [dostęp 2022-06-30] (ang.).
- ↑ B. Erdmann, C. Keller, Actinide(lanthanide)-noble metal alloy phases, preparation and properties, „Journal of Solid State Chemistry”, 7 (1), 1973, s. 40–48, DOI: 10.1016/0022-4596(73)90119-9 [dostęp 2022-06-30] (ang.).
- ↑ C. Keller, L. Koch, K.H. Walter, Die reaktion der transuranoxide mit alkalioxiden—II, „Journal of Inorganic and Nuclear Chemistry”, 27 (6), 1965, s. 1225–1232, DOI: 10.1016/0022-1902(65)80084-7 [dostęp 2022-06-30] (niem.).
- ↑ C. Keller, L. Koch, K.H. Walter, Die reaktion der oxide der transurane mit alkalioxiden—I, „Journal of Inorganic and Nuclear Chemistry”, 27 (6), 1965, s. 1205–1223, DOI: 10.1016/0022-1902(65)80083-5 [dostęp 2022-06-30] (niem.).
- ↑ T. Stchouzkoy, H. Pezerat. Study of the oxides and the peroxide of protactinium. „Physico-Chimie du Protactinium”, s. 61–72, 1966. Paryż: Institut de Recherche Chimique Appliquee.
- ↑ R. Lorenz, H.L. Scherff, N. Toussaint, The carbothermic reduction of protactinium pentoxide and first results on protactinium carbide, „Journal of Inorganic and Nuclear Chemistry”, 31 (8), 1969, s. 2381–2390, DOI: 10.1016/0022-1902(69)80568-3 [dostęp 2022-06-30] (ang.).
- ↑ Protactinium:halides. W: The Chemistry of the Actinide and Transactinide Elements. Dordrecht: Springer Netherlands, 2011, s. 197–204. ISBN 978-94-007-0211-0. (ang.).
- ↑ Protactinium. W: John Emsley: Nature’s Building Blocks: An A-Z Guide to the Elements. Oxford, England, UK: Oxford University Press, 2003-08-11, s. 347–349. ISBN 0-19-850340-7. (ang.).
- ↑ Sven A.E. Johansson, Decay of UX1, UX2, and UZ, „Physical Review”, 96 (4), 1954, s. 1075–1080, DOI: 10.1103/PhysRev.96.1075 [dostęp 2022-06-30] (ang.).
- ↑ Fajans, K. and Gohring, O. Über das Uran X2-das neue Element der Uranreihe. „Physikalische Zeitschrift”. 14, s. 877–884, 1913. (niem.).
- ↑ K. Fajans, O. Göhring, Über die komplexe Nature des UrX, „Die Naturwissenschaften”, 1 (14), 1913, s. 339–339, DOI: 10.1007/BF01495360 [dostęp 2022-06-30] (niem.).
- ↑ W.R. Shea: Otto Hahn and the Rise of Nuclear Physics. New York: Springer-Verlag, 1983, s. 213. ISBN 90-277-1584-X. (ang.).
- ↑ Ryohei Nakamura, Mochida Kyuhei, シンチレータ (Scintillator), patent JP 11166177A, 22 czerwca 1999 (jap.).
- ↑ Yadav Tapesh, Alexander John, Inorganic dopants, inks and related nanotechnology, zgłoszenie patentowe US 20040170820, 2 września 2004 (ang.).
- ↑ K.W. Bagnall, The Chemistry of Weighable Amounts of Polonium and Protactinium, „ract”, 5 (1), 1966, s. 1–5, DOI: 10.1524/ract.1966.5.1.1 [dostęp 2022-06-30] (ang.).
| Układ okresowy pierwiastków | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3[i] | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |||||||||||||||||||||||||||
| 1 | H | He | ||||||||||||||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | ||||||||||||||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | ||||||||||||||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | ||||||||||||||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | ||||||||||||||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | ||||||||||||
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | ||||||||||||
| 8 | Uue | Ubn | ✱ | |||||||||||||||||||||||||||||||||||||||||
| ✱ | Ubu | Ubb | Ubt | Ubq | Ubp | Ubh | Ubs | ...[ii] | ||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||