신생물
 | |
|---|---|
| 다른 이름 | 종양 |
 | |
| 침습성 대장암의 대장 절제술 표본(Colectomy specimen), 상단 중앙에 있는 붉은색으로 불규칙하게 형성된 분화형 종양. | |
| 진료과 | 종양학 |
| 증상 | 덩어리 형성 |
| 합병증 | 암 |
| 병인 | 방사선, 환경 요인, 특정 감염 |
신생물[1](新生物, 영어: neoplasm, 종양)은 조직의 비정상적이고 과도한 성장의 한 유형이다[2][3]. 신생물이 형성되거나 생성되는 과정을 신생물 형성(新生物形成, 영어: neoplasia)이라고 한다. 신생물의 성장은 주변 정상 조직의 성장과 달리 통제되지 않으며, 원래의 유발 요인이 제거되더라도 비정상적으로 계속 성장할 수 있다.[4][5][6] 이러한 비정상적인 성장은 보통 양성 종양이나 악성 종양를 형성하는데, 이 때 덩어리 형태로 이루어질 경우 종양이라고 한다.[7]
ICD-10에서는 신생물을 양성 신생물, 제자리 신생물, 악성 신생물, 그리고 불확실하거나 알려지지 않은 활동을 하는 신생물의 네 가지 주요 그룹으로 분류한다.[8] 악성으로 발전되는 신생물은 흔히 암으로도 칭하며, 종양학의 주요 연구 분야이다.
조직의 비정상적인 성장으로 나타나는 신생물 형성 이전 세포들은 종종 비정상적인 성장 양상을 겪으며, 이는 화생이나 이형성과 같다.[9] 그러나 화생이나 이형성은 항상 신조직 형성으로 진행되지 않을 수 있으며, 신생물 형성은 다른 상황에서도 발생할 수 있다.[4]
종양 관련 용어의 정의
[편집]한글 용어는 KMLE[10]에 준하여 서술
• 역형성(Anaplasia) : 세포 또는 세포 그룹 내의 구조적 분화 손실
• 무형성(Aplasia) : 장기 또는 장기의 일부가 없는 경우
• 섬유조직형성(Desmoplasia) : 결합 조직 성장
• 이형성(Dysplasia) : 세포 또는 조직 표현형의 변화
• 과형성(Hyperplasia) : 세포의 증식
• 형성저하증(Hypoplasia) : 선천적으로 평균 이하의 세포 수, 특히 부적절한 경우
• 화생(Metaplasia) : 세포 유형 전환
• 신생물 형성(Neoplasia) : 비정상적인 증식
• 전진형성(Prosoplasia) : 새로운 세포 기능 개발
• 무활력(Abiotrophy) : 장기 또는 조직의 활력 저하
• 위축(Atrophy) : 세포의 수 또는 부피 감소와 함께 장기의 기능 저하
• 비대(Hypertrophy) : 세포 또는 조직의 부피 증가
• 영양장애위축(Hypotrophy) : 세포 또는 조직의 부피 감소
• 퇴행위축(Dystrophy) : 부적절하거나 잘못된 영양 섭취로 인한 모든 퇴행성 질환
종류
[편집]신생물은 양성 종양, 잠재적 악성 또는 악성(암)일 수 있다.[11]
- 양성 종양에는 자궁근종, 골종양, 멜라닌 세포 모반(피부 점)이 포함된다. 이러한 종양은 주변이 둘러싸고 국한되어 있으며 암으로 변하지 않는다.[12]
- 잠재적 악성 신생물에는 상피내암(제자리암)이 포함된다. 이러한 종양은 국소적이며 침입하거나 파괴하지 않지만 시간이 지나면 암으로 변할 수 있다.
- 악성 신생물은 일반적으로 암이라고 불린다. 악성 신생물은 주변 조직을 침범하여 파괴하고 전이를 일으킬 수 있으며, 치료하지 않거나 치료에 반응하지 않으면 일반적으로 치명적인 결과를 초래한다.
- 이차성 종양은 원발성 종양의 전이성 파생물 또는 화학 요법이나 방사선 요법과 같은 특정 암 치료 후 빈도가 증가하는 모든 종류의 암 종양을 말한다.
- 드물게 원발암 부위가 알려지지 않은 전이성 신생물이 있을 수 있으며, 이는 원발암을 알 수 없는 암으로 분류된다.
클론성
[편집]신생물들은 종종 이질성일 수 있지만, 일반적으로 두 가지 이상의 유형의 세포를 포함하면서도, 그 시작과 지속적인 성장은 단일 클론 세포 집단에 의존한다.[13] 이러한 세포들은 동일한 세포에서 유래하고 모두 동일한 유전적 또는 후성유전학적 이상을 지니고 복제 가능하기 때문에 단일 클론에서 기원하는 것으로 추정된다.
림프종 및 백혈병과 같은 림프성 신생물의 경우, 면역글로불린 유전자(B 세포 병변의 경우) 또는 T 세포 수용체 유전자(T 세포 병변의 경우)의 단일 재배열 증폭을 통해 클론성이 입증된다. 즉, 림프구 세포 증식을 신생물 형성으로 식별하기 위해서는 클론성 입증이 필요하다.[14]
이 외의 경우 클론성 입증이 항상 가능한 것은 아니므로, 신생물을 클론성 세포 증식으로 정의할 수 없다. 또한 마찬가지로 신생물의 정의에서 클론성이 반드시 필요하지는 않다.
신생물과 종양
[편집]단어 '종양' 또는 'tumour'은 부종을 의미하는 라틴어에서 유래하였다. 이 단어는 원래 모든 형태의 부종을 뜻하는 것으로, 신생물이던 아니던 모두를 지칭하였다. 현재는 종양이 신생물(고체 또는 액체로 채워진 낭종으로, 비정상적인 신생세포의 성장에 의해 형성될 수도 있고 아닐 수도 있다)의 동의어로 사용되며, 크기가 커 보이는 것을 특징으로 한다.[15][16] 일부 신생물들은 덩어리를 형성하지 않으며 이에는 백혈병과 대부분의 상피내암이 포함된다. 또한 종양은 암과 동일한 의미를 가지지 않는다. 암은 정의상 악성이지만, 종양은 양성, 상피내암, 또는 악성일 수 있다.
덩어리(Mass)와 결절(nodule)이라는 용어들은 종종 종양(tumor)과 동일한 의미로 사용된다. 일반적으로 말하자면, 덩어리라는 용어가 병변의 크기를 참조하지 않고 일반적으로 사용되나, 좀 더 구체적으로 말하자면 병변 최대 직경이 20mm 이상인 경우에 주로 종양, 병변 크기가 최대 직경 20mm보다 작을 때 주로 결절이라고 표현한다.[17]
원인
[편집]

인간의 종양은 단일 세포 내에 축적된 유전적 및 후성유전학적 변화의 결과로 발생하며, 이로 인해 세포가 통제할 수 없이 분열하고 확장된다.[18] 종양은 유전적 돌연변이로 인해 발생할 수 있는 조직의 비정상적인 증식으로 인해 발생할 수 있다. 그러나 모든 유형의 신생물이 조직의 종양성 과다 성장을 유발하는 것은 아니며(예: 백혈병 또는 상피내암), 신생물의 성장과 일반적인 조직의 재생 과정(예: 분화 및 빠른 세포 증식)은 유사하다.[19]
종양의 성장은 수학과 연속체 역학을 사용하여 연구되어 왔다. 따라서 혈관종이나 림프관종(혈액 또는 림프관에서 형성됨)과 같은 혈관 종양은 끈적끈적한 세포로 형성된 단단한 골격과 세포가 성장할 수 있는 공간을 채우는 유기 액체가 합쳐진 것으로 간주된다.[20] 이러한 유형의 모델에서는 기계적 스트레스와 변형을 다루고 종양의 성장과 주변 조직 및 혈관계에 미치는 영향을 규명할 수 있다. 이 모델을 사용한 실험의 최근 연구 결과에 따르면 종양의 활발한 성장은 종양의 바깥쪽 가장자리로 제한되며, 기저 정상 조직의 경직이 종양 성장도 억제하는 것으로 나타났다.[21]
조직 비정상 증식과 관련 없는 양성 질환인 피지 낭종 등도 악성 가능성 없이 종양으로 나타난다. 임신 중 등 다른 시기에 자주 발견되는 유방 낭종은 다른 캡슐화된 선종(갑상선, 부신, 췌장)과 마찬가지로 종양의 예이다.
캡슐화된 혈종, 캡슐화된 괴사 조직(벌레 물림, 이물질 또는 기타 유해한 기전으로 인한), 켈로이드(흉터 조직의 개별적인 과증식) 및 육아종도 종양으로 나타난다.
유출 폐쇄 또는 협착, 비정상적인 연결 등으로 인해 정상 구조물(요관, 혈관, 간내 또는 간외 담관, 폐 내포물 또는 위장 중복)의 국소적인 비대가 종양으로 나타날 수 있다. 동정맥 누공 또는 혈전증, 담도 순환 장애나 동맥류 등이 그 예이다. 내용물이 유출될 경우 치명적인 결과를 초래할 수 있는 여러 유형의 종양은 생검하는 것이 위험할 수 있다. 이러한 유형의 종양이 발견되면 이러한 심각한 합병증을 피하기 위해 조직 검사나 외과적 탐색/절제 전에 초음파, CT 스캔, MRI, 혈관 조영술, 핵의학 검사 등의 진단 방식을 사용한다.
악성 신생물
[편집]DNA 손상
[편집]
DNA 손상은 암으로 알려진 악성 신생물의 주요 근본 원인으로 간주된다.[22] 이 문단의 그림에서 암으로의 진행에 있어 DNA 손상의 중심적인 역할은 상단 근처의 상자에 설명되어 있다. (암으로의 진행에 있어 DNA 손상, 후성유전학적 변화 및 DNA 복구 결핍의 중심적인 특징은 빨간색으로 표시되어 있다.) DNA 손상은 매우 흔하다. 자연적으로 발생하는 DNA 손상(주로 세포 대사와 체온에서 물속 DNA 의 특성 때문에)은 인간 세포당 하루 평균 60,000 개 이상의 새로운 손상이 발생하는 속도로 발생한다. 외인성 물질에 노출되면 추가적인 DNA 손상이 발생할 수 있다. 담배 연기는 외인성 DNA 손상을 증가시키며 이러한 DNA 손상은 흡연으로 인한 폐암의 원인일 가능성이 높다.[23] 태양 복사에 의한 자외선은 흑색종에서 중요한 DNA 손상을 유발한다.[24] 헬리코박터 파일로리 감염은 높은 수준의 활성산소를 생성하여 DNA를 손상시키고 위암을 유발한다.[25] 고지방 식단을 섭취하는 사람들의 결장에서 고농도의 담즙이 DNA를 다치게 하고 대장암을 일으킨다.[26] Katsurano 등이 저술한 연구에서는 염증이 생긴 대장 상피의 대식세포와 호중구가 대장 종양 형성을 시작하는 DNA 손상을 유발하는 활성 산소 종의 발생 기원이라고 밝혔다.[27] 그림 상단에 있는 상자에는 DNA 손상의 일부 원인이 표시되어 있다.
34개의 DNA 복구 유전자 중 생식선에서 기원하는 돌연변이가 있어 유전자 결함을 유발하는 사람의 경우에는 암에 걸릴 위험이 높다. DNA 복구 유전자의 일부 생식선 돌연변이는 평생 암에 걸릴 확률이 최대 100%에 이른다(예: p53 유전자 돌연변이).[28] 이러한 생식선 기원 돌연변이는 그림에서 왼쪽의 상자에 화살표로 표시되어 있으며, 해당 돌연변이가 DNA 복구 결핍에 기여함을 나타낸다.
악성 신생물의 약 70%는 유전적 요소가 없으며 "산발성 암"으로 불린다.[29] 산발성 암 중 소수만이 DNA 복구 유전자의 돌연변이로 인해 DNA 복구에 결함을 보인다. 그러나 산발성 암 대부분은 후성유전학적 변화로 인해 DNA 복구 유전자 발현을 감소시키거나 침묵시켜, DNA 복구에 결함을 보인다. 예를 들어, 113개의 순차적 대장암 중 단 4개만이 DNA 복구 유전자 MGMT(O-6-methylguanine-DNA methyltransferase)에서 과오 돌연변이를 가지고 있는 반면, 대부분은 MGMT 프로모터 영역의 메틸화(후성유전학적 변형)로 인해 MGMT 발현 감소를 보였다.[30] 5건의 보고서에서는 대장암의 40~90%에서 MGMT 프로모터 영역의 메틸화로 인해 MGMT 발현 감소가 확인되었다.[31][32][33][34][35]
마찬가지로, PMS2 발현 결여를 가진 불일치 복구결핍 대장암 119례 중에서 PMS2 유전자 돌연변이 때문에 PMS2 가 부족한 경우는 6례였고, 프로모터 메틸화 때문에 페어링 파트너인 MLH1 이 억제되어 PMS2 발현이 결핍된 경우는 103례였다(MLH1 이 없을 때 PMS2 단백질의 불안정).[36] 나머지 10건의 경우에는, PMS2 발현결핍은 후성유전학적 과발현으로 인해 MLH1 을 하향 조절하는 마이크로 RNA 인 miR-155 가 과발현되었기 때문일 가능성이 높다.[37]
다른 예에서, 후성유전학적 결함은 13%-100% 사이의 빈도로 DNA 복구 유전자 BRCA1, WRN, FANCB, FANCF, MGMT, MLH1, MSH2, MSH4, ERCC1, XPF, NEIL1 와 ATM.에서 발견되었다. 이러한 후성유전학적 결함은 다양한 암(예: 유방암, 난소암, 대장암, 두경부암)에서 발생했다. Facista 등이 평가한 49개 대장암의 대부분에서 ERCC1, XPF 또는 PMS2의 발현에 두세 가지 결함이 동시에 나타났다.[38] 이 문단의 그림 상단에서 세 번째 레벨의 중앙 상자에 DNA 복구 유전자의 발현 감소를 유발하는 후성유전학적 변화가 표시되어 있고 네 번째 레벨에 그에 따른 DNA 복구 결함이 표시되어 있다. DNA 복구 유전자의 발현이 감소하면 DNA 손상이 정상보다 높은 수준으로 세포에 축적되고 이러한 과도한 손상으로 인해 돌연변이나 돌연변이 발생 빈도가 증가한다. DNA 불일치 복구[39][40]나 상동 재조합 복구(HRR)[41]에 결함이 있는 세포에서는 돌연변이 발생률이 크게 증가한다.
DNA 이중 가닥 파손을 복구하거나 다른 DNA 손상을 복구하는 동안 복구 부위가 제대로 제거되지 않으면 후성유전학적 유전자 침묵을 일으킬 수 있다.[42][43] DNA 복구 결핍(그림의 4 단계)은 DNA 손상(그림의 5 단계)을 증가시켜 체세포 돌연변이와 후성유전학적 변형을 증가시킨다(그림의 6 단계). 정상처럼 보일 수 있는 조직 내부에서도 여러 가지 변형들로 인해 구역 결함이 발생하며, 이는 악성 신생물에서 무질서하고 부적절한 증식을 보이는 복제 조직의 발생에 대한 일반적인 전구체가 된다. 이러한 구역 결함(그림 하단에서 두 번째 수준)에는 여러 돌연변이와 후성유전학적 변화가 있을 수 있다.
일단 암이 형성되면 일반적으로 게놈은 불안정해진다. 이러한 불안정성은 DNA 복구 감소나 과도한 DNA 손상 때문일 수 있다. 이러한 불안정성으로 인해 암은 계속 진화하고 하위 클론을 생성한다. 예를 들어, 9개 부위에서 채취된 신장암 샘플에는 40개의 유비퀴틴 돌연변이가 있어 종양의 이질성(즉, 암의 모든 부위에 존재)을 보여주었다. 그리고 59개의 돌연변이는 일부(모든 부위는 아님)에서 공유했다. 반면, 29개의 "개별" 돌연변이는 암의 한 부위에만 존재했다.[44]
구역 결함(field defect)
[편집]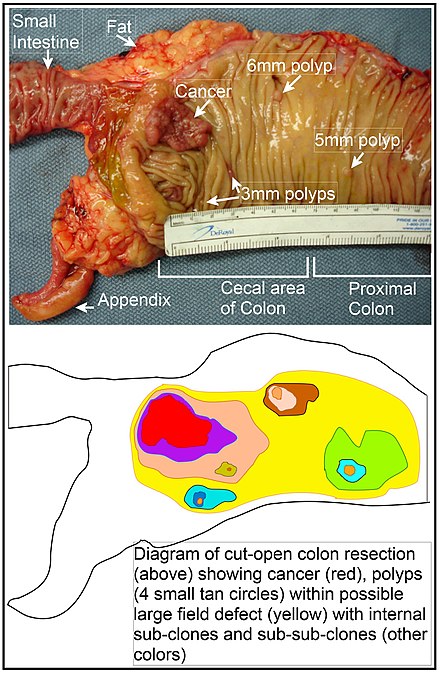
이 현상을 설명하기 위해 "구역 효과(field effect)", "구역 암화(field cancerization)", "구역 발암(field carcinogenesis)" 등 다양한 다른 용어가 사용되었다. "구역 암화"라는 용어는 1953년에 암이 발생하기 쉽도록 당시에는 거의 알려지지 않은 과정에 의해 전제 조건이 형성된 상피 영역 또는 "구역"을 설명하기 위해 처음 사용되었다.[45] 그 이후로 "구역 암화"및 "구역 결함"이라는 용어는 새로운 암이 발생할 가능성이 있는 전 악성 조직을 설명하는 데 사용되었다.
구역 결함은 암으로의 진행에 중요하다.[46][47] 그러나 대부분의 암 연구에서 Rubin[48]이 지적한 것처럼, 암 연구의 대부분은 생체 내에서 잘 정의된 종양 또는 시험관 내에서 개별 종양 병소에 대해 수행되었다. 그러나 돌연변이 표현형 인간 대장 종양에서 발견된 체세포 돌연변이의 80% 이상은 말기 클론 확장 시작 전에 이미 있던 것으로 밝혀졌다.[49] 마찬가지로, Vogelstein 등[50]은 종양에서 확인된 체세포 돌연변수 중 절반 이상이 정상세포가 성장하는 동안 종양 전 단계(전장결함) 에서 일어난 것임을 지적하였다. 유사하게, 종양에 존재하는 후성유전학적 변화는 종양 전 구역 결함에서 발생했을 수 있다.
구역 효과에 대한 확장된 관점은 종양 전 세포의 분자 및 병리학적 변화뿐만 아니라 종양 시작부터 환자 사망까지 종양 진화에 대한 외인성 환경 요인과 국소 미세 환경의 분자 변화의 영향을 포괄하는 "병인성 구역 효과"라고 불린다.[51] 결장에서 구역 결함은 결장 안쪽 표면에 있는 장의 선와 중 하나의 기저에 있는 줄기세포 중 돌연변이 또는 후성유전학적으로 변형된 세포의 자연 선택에 의해 발생할 수 있다. 돌연변이 또는 후성유전학적으로 변형된 줄기세포는 자연 선택에 의해 주변의 다른 줄기세포를 대체할 수 있으므로, 비정상적인 조직이 발생할 수 있다. 이 문단의 그림에는 대장암과 4개 용종을 보여주는 절제 후 세로로 절개한 대장 절편 사진이 포함되어 있다. 사진 아래에서 돌연변이 또는 후성유전학적으로 변형된 세포가 어떻게 큰 클론을 형성하는지 다이어그램이 나와 있으며, 그림에서 노란색으로 표시된 넓은 영역이 그 예시이다.
그림의 첫 번째 큰 클론(세포의 큰 클론) 내에서 두 번째 돌연변이 또는 후성유전학적 변경이 발생하여 특정 줄기세포가 단편 내의 다른 줄기세포에 비해 이점을 얻을 수 있었으며, 이렇게 변경된 줄기세포는 복제를 통해 확장되어 원래 클론 내에서 2차 클론 또는 하위 클론(Subclone)을 형성할 수 있었다. 이는 다이어그램에서 큰 노란색 원본 영역 내에 서로 다른 바탕색의 작은 클론 4개로 표시되었다. 이러한 새로운 클론(하위 클론) 내에서, 이 과정은 작은 용종 또는 악성 신생물(암)을 생성하는 줄기세포가 발생할 때까지 복제적으로 확장되는 4개의 추가 보조 클론(다이어그램에서 다른 색상, Sub-Subclone) 내의 작은 클론으로 표시된 것처럼 여러 번 반복될 수 있다.
사진에서 결장의 이 부분에 명백한 구역 결함으로 인해 4개의 용종(용종의 크기가 6mm, 5mm, 3mm 2개로 표시됨)과 가장 긴 크기가 약 3cm인 암이 생겼다(암의 크기가 각각 6mm, 5mm, 3mm 로 표시됨). 이러한 신생물들은 사진 아래 다이어그램에서도 각각 네 개의 작은 황갈색 원(용종)과 하나의 더 큰 빨간색 영역 (암)으로 표현되었다. 사진 속 암은 결장이 소장과 연결되는 부위와 충수가 발생하는 부위인 맹장 부위에서 발견되었다. 사진 속 지방 조직들은 결장 외벽 외부에 위치하고 있다. 여기에 나타난 결장 부분은 세로 방향으로 절단하여 그 안쪽 표면을 드러내고, 결장 내부 상피 내벽에서 발생하는 암과 용종을 표시하였다.
산발성 대장암이 발생하는 일반적인 과정이 자연 선택에 의해 퍼지는 전 신생물 클론의 형성에 이어 초기 클론 내에 내부 하위 클론과 그 내부의 하위 하위 클론이 형성되는 것이라면, 대장암은 일반적으로 전 악성 사건의 연속을 반영하는 증가하는 이상 영역과 관련이 있어야 하며 그 선행이 되어야 한다. 가장 광범위한 이상 영역 (다이어그램에서 가장 바깥 쪽 노란색 불규칙한 영역)은 악성 신생물 형성의 가장 초기 사건을 반영한다.
암의 특정 DNA 복구 결핍에 대한 실험적 평가에서, 많은 특정 DNA 복구 결핍은 해당 암을 둘러싼 구역 결함에서도 발생하는 것으로 나타났다. 아래 표에는 암의 DNA 복구 결핍이 후성유전학적 변화로 인한 것으로 밝혀진 사례와 후성유전학적으로 동일한 DNA 복구 결핍이 주변 구역 결함에서 발견되는 다소 낮은 빈도의 예시가 나와 있다.
산발성 암 및 인접 분야 결함에서 DNA 복구 유전자의 후성유전학적 변화 빈도
| 암종 | 유전자 | 암 발생 빈도 | 구역 결함 빈도 | 출처 |
|---|---|---|---|---|
| 대장 | MGMT | 46% | 34% | [52] |
| 대장 | MGMT | 47% | 11% | [53] |
| 대장 | MGMT | 70% | 60% | [54] |
| 대장 | MSH2 | 13% | 5% | [53] |
| 대장 | ERCC1 | 100% | 40% | [55] |
| 대장 | PMS2 | 88% | 50% | [55] |
| 대장 | XPF | 55% | 40% | [55] |
| 두경부 | MGMT | 54% | 38% | [56] |
| 두경부 | MLH1 | 33% | 25% | [57] |
| 두경부 | MLH1 | 31% | 20% | [58] |
| 위 | MGMT | 88% | 78% | [59] |
| 위 | MLH1 | 73% | 20% | [60] |
| 식도 | MLH1 | 77%-100% | 23%-79% | [61] |
열린 결장 부분 사진에 보이는 구역 결함의 작은 용종 중 일부는 비교적 양성 종양일 수 있었다. 대장내시경 검사에서 발견되어 3년 동안 반복 대장내시경 검사를 받은 10mm 미만의 용종 중 25%는 크기가 변하지 않았고, 35%는 퇴행하거나 크기가 줄어든 반면, 40%는 크기가 커진 것으로 나타났다.[62]
게놈 불안정성(Genome instability)
[편집]암은 게놈 불안정성(genome instability) 또는 돌연변이 표현형(mutator phenotype)을 나타내는 것으로 알려져 있다.[63] 핵 내의 단백질 코딩 DNA는 전체 게놈 DNA의 약 1.5%였다.[64] 이 단백질 코딩 DNA(진유전체) 내에서 유방암이나 대장암은 평균적으로 약 60~70개의 단백질 변이 돌연변이를 가질 수 있으며, 이 중 약 34개는 "운전자" 돌연변이고 나머지는 "승객" 돌연변이다.[65] 그러나 유방암 조직 샘플 내 전체 게놈(비단백질 코딩 영역 등)의 평균 DNA 서열 돌연변이 수는 약 20,000개였다.[66] 평균 흑색종 조직 샘플(흑색종의 진유전체 돌연변수 빈도가 더 높은 경우[65])에서 전체 DNA 서열 돌연변수 수가 약 8만 개[67]로 확인되어 인간 세대 간 (부모와 자식 간) 전체 게놈에서 발생하는 매우 낮은 (70개 정도)돌연변이 수 비율과 차이가 났다.[68][69]
전체 염기서열에 걸친 변화량을 고려한다면 초기 구역 결함 형성이 암을 유발하는 구역 결함의 초기 변화(예: 이 문단의 다이어그램에서 노란색 영역)가 DNA 복구 결함이라는 것을 시사한다. Facista 등[70]에 따르면 대장암을 둘러싼 큰 구역 결함(암의 양쪽에서 약 10cm까지 확장)은 구역 결함의 전체 영역에서 2~3개의 DNA 복구 단백질(ERCC1, XPF 또는 PMS2)에 후성유전학적 결함이 자주 발생하는 것으로 나타났다. DNA 복구 결핍은 돌연변이 발생률을 증가시킨다.[71][72][73] DNA 복구에 문제가 있으면 그 자체로 DNA 손상이 누적될 수 있으며, 이러한 손상 중 일부를 지나치게 많이 전사하거나 합성하는 등의 오류를 일으켜 돌연변수를 일으킬 수 있다. 또한 이렇게 축적된 DNA 손상을 제대로 복구하지 못하면 돌연변이가 발생할 수 있다. 이러한 새로운 돌연변이는 증식 이점을 제공하여 구역 결함을 생성할 수 있었다. DNA 복구 유전자의 돌연변이 / 신돌연변이 (mutations/epimutations)는 그 자체로는 선택적 이점을 부여하지 않지만 세포가 증식 이점을 제공하는 추가 돌연변이 / 신돌연변이 (additional mutations/epimutations)를 획득할 때 세포 증식의 승객으로 운반될 수 있다.
용어
[편집]신생물(Neoplasia)이라는 용어는 종양의 동의어이다. 신생물 형성(Neoplasia)은 신생물/종양의 형성 과정을 나타내며 그 과정을 신생물 형성이라고 한다. 신생물(Neoplasm)이라는 단어 자체는 그리스어 neo (새로운)와 plastic (형성, 성형)에서 유래했다.
종양은 라틴어 명사 tumor (부종), 최종적으로 동사에서 파생된 tumere (부풀다)에서 왔다. 영연방에서는 철자를 tumour 로 일반적으로 사용하는 반면 미국에서는 일반적으로 철자를 tumor 로 사용한다. 전통적인 의학적인 의미에서 종양은 살이 비정상적으로 부어오르는 것을 의미했다. 로마의 의학 백과사전 저술가 셀수스(기원전 30~38년경)는 급성 염증의 네 가지 주요 징후를 tumor, dolor, calor, rubor (부종, 통증, 열감, 발적)로 설명하였다. (그의 저서인 De Medicina은 이동식 인쇄기가 발명된 후 1478년에 인쇄된 최초의 의학 서적이었다.)
현대 영어에서 종양이라는 단어는 종종 낭성(액체로 채워진) 성장 또는 고형 종양(암성 또는 비암성)의 동의어로 사용되며[74], 다른 형태의 부종은 보통 "붓기"나 "팽창" 등으로 불린다.[75]
관련 용어인 명사 tumescence (종창)과 tumefaction (종괴)는 형용사 tumescent 에서 파생된 것으로, 종창과 종괴는 종양성 세포가 아닌 다른 원인에 의한 증식을 설명하는 현재 의학 용어이다.[76] 이런 유형인 종창은 외상, 감염 및 기타 요인으로 인한 염증으로 인해 발생하는 경우가 많다.
종양은 종양 세포의 과다 증식 이외의 다른 질환으로 인해 발생할 수도 있다. 낭종(예: 피지 낭종)은 종양 세포가 없더라도 종이라고 부르기도 한다. 이는 병리학적 진단이 아직 내려지지 않은 증식을 가리킬 때 사용한다.
같이 보기
[편집]각주
[편집]- ↑ 대한의협 의학용어 사전 https://www.kmle.co.kr/search.php?Search=neoplasm&EbookTerminology=YES&DictAll=YES&DictAbbreviationAll=YES&DictDefAll=YES&DictNownuri=YES&DictWordNet=YES
- ↑ 〈neoplasm〉. 《Lexico UK English Dictionary》. Oxford University Press. 2021년 4월 28일에 원본 문서에서 보존된 문서.
- ↑ “neoplasm”. 《Dictionary.com》. 랜덤하우스.
- ↑ 가 나 Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (July 2014). “Type-2 pericytes participate in normal and tumoral angiogenesis”. 《Am. J. Physiol., Cell Physiol.》 307 (1): C25–38. doi:10.1152/ajpcell.00084.2014. PMC 4080181. PMID 24788248.
- ↑ Cooper GM (1992). 《Elements of human cancer》. Boston: Jones and Bartlett Publishers. 16쪽. ISBN 978-0-86720-191-8.
- ↑ Taylor, Elizabeth J. (2000). 《Dorland's Illustrated medical dictionary.》 29판. Philadelphia: Saunders. 1184쪽. ISBN 978-0721662541.
- ↑ 《Stedman's medical dictionary》 28판. Philadelphia: Lippincott Williams & Wilkins. 2006. Neoplasm쪽. ISBN 978-0781733908.
- ↑ “II Neoplasms”. 《International Statistical Classification of Diseases and Related Health Problems 10th Revision (ICD-10) Version for 2010》. World Health Organization. 2018년 7월 24일에 원본 문서에서 보존된 문서. 2014년 6월 19일에 확인함.
- ↑ Abrams, Gerald. “Neoplasia I”. 2015년 10월 31일에 원본 문서에서 보존된 문서. 2012년 1월 23일에 확인함.
- ↑ “KMLE”.
- ↑ “Cancer - Activity 1 - Glossary, page 4 of 5”. 2008년 5월 9일에 원본 문서에서 보존된 문서. 2008년 1월 8일에 확인함.
- ↑ Abrams, Gerald. “Neoplasia I”. 2015년 10월 31일에 원본 문서에서 보존된 문서. 2012년 1월 23일에 확인함.
- ↑ “Medical Definition of Clone”. 2012년 10월 25일에 원본 문서에서 보존된 문서. 2015년 2월 10일에 확인함.
- ↑ Lee ES, Locker J, Nalesnik M, Reyes J, Jaffe R, Alashari M, Nour B, Tzakis A, Dickman PS (January 1995). “The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation”. 《N. Engl. J. Med.》 332 (1): 19–25. doi:10.1056/NEJM199501053320104. PMID 7990861.
- ↑ “Pancreas Cancer: Glossary of Terms”. 2010년 6월 5일에 원본 문서에서 보존된 문서. 2008년 1월 8일에 확인함.
- ↑ 〈Tumor〉. 《Dorland's Illustrated Medical Dictionary》 31판. Saunders. 2007. ISBN 978-1-84972-348-0.
- ↑ Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (July 2014). “Type-2 pericytes participate in normal and tumoral angiogenesis”. 《Am. J. Physiol., Cell Physiol.》 307 (1): C25–38. doi:10.1152/ajpcell.00084.2014. PMC 4080181. PMID 24788248.
- ↑ Tammela, Tuomas; Sage, Julien (2020). “Investigating Tumor Heterogeneity in Mouse Models”. 《Annual Review of Cancer Biology》 4 (1): 99–119. doi:10.1146/annurev-cancerbio-030419-033413. PMC 8218894. PMID 34164589.
- ↑ Asashima M, Oinuma T, Meyer-Rochow VB (1987). “Tumors in amphibia”. 《Zoological Science》 4: 411–425.
- ↑ Ambrosi D, Mollica F (2002). “On the mechanics of a growing tumor”. 《International Journal of Engineering Science》 40 (12): 1297–316. doi:10.1016/S0020-7225(02)00014-9.
- ↑ Volokh KY (September 2006). “Stresses in growing soft tissues”. 《Acta Biomater》 2 (5): 493–504. doi:10.1016/j.actbio.2006.04.002. PMID 16793355.
- ↑ Kastan MB (2008). “DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture”. 《Mol. Cancer Res.》 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- ↑ Cunningham FH, Fiebelkorn S, Johnson M, Meredith C (November 2011). “A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants”. 《Food Chem. Toxicol.》 49 (11): 2921–33. doi:10.1016/j.fct.2011.07.019. PMID 21802474.
- ↑ Kanavy HE, Gerstenblith MR (December 2011). “Ultraviolet radiation and melanoma”. 《Semin Cutan Med Surg》 30 (4): 222–8. doi:10.1016/j.sder.2011.08.003. PMID 22123420.
- ↑ Handa O, Naito Y, Yoshikawa T (2011). “Redox biology and gastric carcinogenesis: the role of Helicobacter pylori”. 《Redox Rep.》 16 (1): 1–7. doi:10.1179/174329211X12968219310756. PMC 6837368. PMID 21605492.
- ↑ Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H (August 2011). “Carcinogenicity of deoxycholate, a secondary bile acid”. 《Arch. Toxicol.》 85 (8): 863–71. doi:10.1007/s00204-011-0648-7. PMC 3149672. PMID 21267546.
- ↑ Katsurano M, Niwa T, Yasui Y, Shigematsu Y, Yamashita S, Takeshima H, Lee MS, Kim YJ, Tanaka T, Ushijima T (January 2012). “Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T- and B-cells in DNA methylation induction”. 《Oncogene》 31 (3): 342–51. doi:10.1038/onc.2011.241. PMID 21685942.
- ↑ Malkin D (April 2011). “Li-fraumeni syndrome”. 《Genes Cancer》 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- ↑ Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (July 2000). “Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland”. 《N. Engl. J. Med.》 343 (2): 78–85. doi:10.1056/NEJM200007133430201. PMID 10891514.
- ↑ Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). “O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions”. 《Gut》 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- ↑ Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (September 2005). “MGMT promoter methylation and field defect in sporadic colorectal cancer”. 《J. Natl. Cancer Inst.》 97 (18): 1330–8. doi:10.1093/jnci/dji275. PMID 16174854.
- ↑ Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G (July 2010). “Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas”. 《World J. Gastroenterol.》 16 (28): 3553–60. doi:10.3748/wjg.v16.i28.3553. PMC 2909555. PMID 20653064.
- ↑ Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH (October 2011). “Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence”. 《Langenbecks Arch Surg》 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233. S2CID 8069716.
- ↑ Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, Cassingena A, Rusconi F, Esposito A, Nichelatti M, Esteller M, Siena S (April 2013). “Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer”. 《Clin. Cancer Res.》 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- ↑ Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, 외. (May 2013). “Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer”. 《Mol. Biol. Rep.》 40 (5): 3851–7. doi:10.1007/s11033-012-2465-3. PMID 23271133. S2CID 18733871.
- ↑ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, 외. (May 2005). “Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer”. 《Gastroenterology》 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ↑ Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, 외. (April 2010). “Modulation of mismatch repair and genomic stability by miR-155”. 《Proc. Natl. Acad. Sci. U.S.A.》 107 (15): 6982–7. Bibcode:2010PNAS..107.6982V. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- ↑ Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, 외. (2012). “Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer”. 《Genome Integr》 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ↑ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). “Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2”. 《Proc. Natl. Acad. Sci. U.S.A.》 94 (7): 3122–7. Bibcode:1997PNAS...94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ↑ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). “Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6”. 《Carcinogenesis》 27 (12): 2402–8. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ↑ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). “Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation”. 《EMBO Rep.》 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- ↑ O'Hagan HM, Mohammad HP, Baylin SB (2008). Lee JT, 편집. “Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island”. 《PLOS Genet.》 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ↑ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV (July 2007). “DNA damage, homology-directed repair, and DNA methylation”. 《PLOS Genet.》 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ↑ Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, 외. (March 2012). “Intratumor heterogeneity and branched evolution revealed by multiregion sequencing”. 《N. Engl. J. Med.》 366 (10): 883–92. doi:10.1056/NEJMoa1113205. PMC 4878653. PMID 22397650.
- ↑ Slaughter DP, Southwick HW, Smejkal W (September 1953). “Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin”. 《Cancer》 6 (5): 963–8. doi:10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. PMID 13094644. S2CID 6736946.
- ↑ Bernstein C, Bernstein H, Payne CM, Dvorak K, Garewal H (February 2008). “Field defects in progression to gastrointestinal tract cancers”. 《Cancer Lett.》 260 (1–2): 1–10. doi:10.1016/j.canlet.2007.11.027. PMC 2744582. PMID 18164807.
- ↑ Nguyen H, Loustaunau C, Facista A, Ramsey L, Hassounah N, Taylor H, Krouse R, Payne CM, Tsikitis VL, Goldschmid S, Banerjee B, Perini RF, Bernstein C (2010). “Deficient Pms2, ERCC1, Ku86, CcOI in field defects during progression to colon cancer”. 《J Vis Exp》 (41): 1931. doi:10.3791/1931. PMC 3149991. PMID 20689513.
- ↑ Rubin H (March 2011). “Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture”. 《BioEssays》 33 (3): 224–31. doi:10.1002/bies.201000067. PMID 21254148. S2CID 44981539.
- ↑ Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, Tavaré S, Shibata D (February 2000). “Genetic reconstruction of individual colorectal tumor histories”. 《Proc. Natl. Acad. Sci. U.S.A.》 97 (3): 1236–41. Bibcode:2000PNAS...97.1236T. doi:10.1073/pnas.97.3.1236. PMC 15581. PMID 10655514.
- ↑ Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). “Cancer genome landscapes”. 《Science》 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- ↑ Lochhead P, Chan AT, Nishihara R, Fuchs CS, Beck AH, Giovannucci E, Ogino S (2014). “Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression”. 《Mod Pathol》 28 (1): 14–29. doi:10.1038/modpathol.2014.81. PMC 4265316. PMID 24925058.
- ↑ Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (September 2005). “MGMT promoter methylation and field defect in sporadic colorectal cancer”. 《J. Natl. Cancer Inst.》 97 (18): 1330–8. doi:10.1093/jnci/dji275. PMID 16174854.
- ↑ 가 나 Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH (October 2011). “Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence”. 《Langenbecks Arch Surg》 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233. S2CID 8069716.
- ↑ Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, 외. (November 2010). “Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers”. 《Gut》 59 (11): 1516–26. doi:10.1136/gut.2009.194787. PMID 20947886. S2CID 206950452.
- ↑ 가 나 다 Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, 외. (2012). “Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer”. 《Genome Integr》 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ↑ Paluszczak J, Misiak P, Wierzbicka M, Woźniak A, Baer-Dubowska W (February 2011). “Frequent hypermethylation of DAPK, RARbeta, MGMT, RASSF1A and FHIT in laryngeal squamous cell carcinomas and adjacent normal mucosa”. 《Oral Oncol.》 47 (2): 104–7. doi:10.1016/j.oraloncology.2010.11.006. PMID 21147548.
- ↑ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, Kokoska MS, Fan CY (October 2009). “Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma”. 《Otolaryngol Head Neck Surg》 141 (4): 484–90. doi:10.1016/j.otohns.2009.07.007. PMID 19786217. S2CID 8357370.
- ↑ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). “Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene”. 《Am J Otolaryngol》 32 (6): 528–36. doi:10.1016/j.amjoto.2010.11.005. PMID 21353335.
- ↑ Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ (November 2009). “Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions”. 《Hum. Pathol.》 40 (11): 1534–42. doi:10.1016/j.humpath.2009.01.029. PMID 19695681.
- ↑ Wani M, Afroze D, Makhdoomi M, Hamid I, Wani B, Bhat G, Wani R, Wani K (2012). “Promoter methylation status of DNA repair gene (hMLH1) in gastric carcinoma patients of the Kashmir valley”. 《Asian Pac. J. Cancer Prev.》 13 (8): 4177–81. doi:10.7314/APJCP.2012.13.8.4177. PMID 23098428.
- ↑ Agarwal A, Polineni R, Hussein Z, Vigoda I, Bhagat TD, Bhattacharyya S, Maitra A, Verma A (2012). “Role of epigenetic alterations in the pathogenesis of Barrett's esophagus and esophageal adenocarcinoma”. 《Int J Clin Exp Pathol》 5 (5): 382–96. PMC 3396065. PMID 22808291.
- ↑ Hofstad B, Vatn MH, Andersen SN, Huitfeldt HS, Rognum T, Larsen S, Osnes M (September 1996). “Growth of colorectal polyps: redetection and evaluation of unresected polyps for a period of three years”. 《Gut》 39 (3): 449–56. doi:10.1136/gut.39.3.449. PMC 1383355. PMID 8949653.
- ↑ Schmitt MW, Prindle MJ, Loeb LA (September 2012). “Implications of genetic heterogeneity in cancer”. 《Ann. N. Y. Acad. Sci.》 1267 (1): 110–6. Bibcode:2012NYASA1267..110S. doi:10.1111/j.1749-6632.2012.06590.x. PMC 3674777. PMID 22954224.
- ↑ Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, 외. (February 2001). “Initial sequencing and analysis of the human genome” (PDF). 《Nature》 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011. 2020년 7월 29일에 원본 문서 (PDF)에서 보존된 문서. 2019년 9월 2일에 확인함.
- ↑ 가 나 Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). “Cancer genome landscapes”. 《Science》 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- ↑ Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, Voest E, Pierce JP, Messer K, Parker BA, Harismendy O, Frazer KA (August 2012). “Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens”. 《Nucleic Acids Res.》 40 (14): e107. doi:10.1093/nar/gks299. PMC 3413110. PMID 22492626.
- ↑ Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, 외. (May 2012). “Melanoma genome sequencing reveals frequent PREX2 mutations”. 《Nature》 485 (7399): 502–6. Bibcode:2012Natur.485..502B. doi:10.1038/nature11071. PMC 3367798. PMID 22622578.
- ↑ Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, 외. (April 2010). “Analysis of genetic inheritance in a family quartet by whole-genome sequencing”. 《Science》 328 (5978): 636–9. Bibcode:2010Sci...328..636R. doi:10.1126/science.1186802. PMC 3037280. PMID 20220176.
- ↑ Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, 외. (November 2012). “Estimating the human mutation rate using autozygosity in a founder population”. 《Nat. Genet.》 44 (11): 1277–81. doi:10.1038/ng.2418. PMC 3483378. PMID 23001126.
- ↑ Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, 외. (2012). “Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer”. 《Genome Integr》 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ↑ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). “Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2”. 《Proc. Natl. Acad. Sci. U.S.A.》 94 (7): 3122–7. Bibcode:1997PNAS...94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ↑ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). “Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6”. 《Carcinogenesis》 27 (12): 2402–8. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ↑ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). “Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation”. 《EMBO Rep.》 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- ↑ Tumor in Medical Encyclopedia
- ↑ 〈Swelling〉. 《MedlinePlus Medical Encyclopedia》. 2012년 10월 14일.
- ↑ 〈tumescence〉. 《옥스퍼드 영어사전》 온라인판. 옥스퍼드 대학교 출판부. (구독 또는 참여 기관 회원가입 필요)