Estrutura das proteínas


A estrutura das proteínas describe os varios niveis de organización das moléculas de proteínas. Refírese ao conxunto de propiedades de disposición no espazo das moléculas de proteína, as cales proceden en último extremo da súa secuencia de aminoácidos, e das características físicas do medio que as rodea e a presenza de compostos simples ou complexos que as estabilicen e/ou induzan nelas un pregamento específico, distinto do esperado espontaneamente. Por tanto, deriva dos seus compoñentes, é dicir, da propia estrutura dos aminoácidos concretos que as compoñen, de como interaccionan quimicamente estes, de forma xerarquizada e específica, e está relacionada co destino celular e a función biolóxica que exercen.
Termodinámica do pregamento
[editar | editar a fonte]En condicións fisiolóxicas, o proceso de pregamento dunha proteína globular está claramente favorecido termodinamicamente, é dicir, o incremento de enerxía libre global do proceso debe ser negativo (aínda que algún dos seus pasos pode ser positivo). Este cambio atínguese equilibrando unha serie de factores termodinámicos como a entropía conformacional, as interaccións carga-carga, as pontes de hidróxeno internas, as interaccións hidrófobas e as interaccións de van der Waals.[1] .[2]
Entropía conformacional
[editar | editar a fonte]Defínese entropía conformacional do pregamento como a diminución da entropía (da aleatoriedade) durante o paso desde unha multitude de conformacións de nobelo aleatorio ata unha única estrutura pregada. A enerxía libre, representada na ecuación ΔG = ΔH - T ΔS, demostra que o ΔS negativo realiza unha contribución positiva a ΔG. É dicir, o cambio de entropía conformacional oponse ao pregamento. Por tanto, o ΔG global, que debe ser negativo, débese a que ou ben ΔH é negativo e grande ou a algún outro aumento da entropía co pregamento. Na práctica danse ambas as cousas.[2]
A orixe do ΔH negativo é o cúmulo de interaccións favorables enerxeticamente que se dan no interior do glóbulo proteico, interaccións que adoitan a non ser covalentes.
Interaccións entre cargas
[editar | editar a fonte]- Artigo principal: Enlace iónico.
As interaccións carga-carga danse entre grupos polares e cargados das cadeas laterais dos aminoácidos compoñentes do polipéptido, xa que os grupos carboxilo e amino do carbono alfa están implicados no enlace peptídico e non interveñen nelas. Deste modo, ditos grupos ionizados atráense e forman un equivalente ao que serían sales entre os residuos do polipéptido: de feito, denomínanse ás veces pontes salinas.[2] Ditas interaccións desaparecen cando o pH do medio fai que se perda o estado de ionización do grupo, e esta é unha das causas da desnaturalización rápida das proteínas cando se engaden ao medio ácidos ou bases, o que fai que as proteínas deban estar nun medio cun pH tamponado e moderado, que é o fisiolóxico, agás excepcións, como pode ser, por exemplo, o interior do lisosoma.[3]
Enlaces de hidróxeno internos
[editar | editar a fonte]- Artigo principal: Enlace de hidróxeno.

As cadeas laterais de moitos aminoácidos compórtanse como doadores ou como aceptores de enlaces de hidróxeno (por exemplo, no caso dos grupos hidroxilo da serina e os grupos amino da glutamina, por exemplo). Ademais, se os protóns das amidas ou os carbonilos do armazón polipeptídico non están implicados no enlace peptídico, poden interaccionar tamén neste tipo de unións estabilizadoras.
Aínda que os enlaces de hidróxeno son febles en disolución acuosa,[2] o seu gran número pode estabilizar a estrutura terciaria das proteínas.
Interaccións de van der Waals
[editar | editar a fonte]- Artigo principal: Forzas de Van der Waals.
O denso empaquetamento no núcleo das proteínas globulares facilita a interacción feble entre grupos moleculares sen carga. Ditos enlaces son de baixa enerxía, mais o seu abundante número compensa a súa debilidade. Cada interacción individual só contribúe nuns poucos kilojoules á entalpía de interacción negativa global. Pero a suma de todas as contribucións de todas as interaccións si que pode estabilizar a estrutura pregada. Deste xeito, unha contribución enerxética favorable feita pola suma das interaccións intramoleculares compensa de modo máis que suficiente a entropía desfavorable do pregamento.[2]
Interaccións hidrófobas
[editar | editar a fonte]- Artigo principal: Interacción hidrófoba.
Por definición, calquera substancia hidrófoba en contacto coa auga provoca que esta se afaste e se agrupe en estruturas denominadas clatratos.[3] Esta ordenación corresponde a unha diminución da entropía do sistema. No caso das proteínas, os residuos hidrófobos dos aminoácidos quedan orientados, no seu pregamento, cara ao interior da molécula, en contacto con outros residuos semellantes e lonxe da auga. En consecuencia, a disposición cara ao interior dos grupos hidrófobos aumenta a aleatoriedade do sistema "proteína máis auga" e, por tanto, produce un aumento de entropía ao dobrarse. Este aumento de entropía produce unha contribución negativa á enerxía libre do pregamento e aumenta a estabilidade da estrutura proteica.[2]
Función das pontes disulfuro
[editar | editar a fonte]- Artigo principal: Ponte disulfuro.

A estabilización da proteína recentemente pregada pode acontecer por medio da formación de pontes disulfuro entre residuos do aminoácido cisteína adxacentes ou enfrontados. Dito procesamento prodúcese en moitos casos no lume do retículo endoplasmático por acción do encima disulfuro isomerase, presente en todas as células eucariotas. Dito encima, que cataliza a oxidación dos grupos sulfhidrilo ou grupos tiol (-SH2) dos residuos de cisteína, é especialmente abondosa en órganos como o fígado ou páncreas, onde se producen pequenas cantidades de proteínas que conteñen este tipo de enlaces.[4]
Niveis de estruturación
[editar | editar a fonte]
A estrutura das proteínas pode xerarquizarse nunha serie de niveis, interdependentes. Estes niveis corresponden a catro estruturas:
- Estrutura primaria, que corresponde á secuencia de aminoácidos.
- Estrutura secundaria, que provoca a aparición de motivos estruturais.
- Estrutura terciaria, que define a estrutura das proteínas compostas por un só polipéptido.
- Estrutura cuaternaria, cando intervén máis dun polipéptido.
Estrutura primaria
[editar | editar a fonte]- Artigo principal: Estrutura primaria das proteínas.
A estrutura primaria das proteínas refírese á secuencia de aminoácidos, é dicir, a combinación liñal dos aminoácidos por medio dun tipo de enlace covalente, o enlace peptídico. Os aminoácidos están unidos por enlaces peptídicos, e unha das súas características máis importante é a coplanaridade dos radicais constituíntes do enlace.
A estrutura liñal do péptido definirá en grande medida as propiedades de niveis de organización superiores da proteína. Esta orde é consecuencia da información do material xenético: Cando se produce a tradución da información contida no ARN (copia complementaria dun xene de ADN) obtense a orde de aminoácidos que van dar lugar á proteína. Pódese dicir, por tanto, que a estrutura primaria das proteínas non é máis que a orde dos aminoácidos que as conforman.
Estrutura secundaria
[editar | editar a fonte]- Artigo principal: Estrutura secundaria das proteínas.
A estrutura secundaria das proteínas é a disposición espacial local do esqueleto proteico, grazas á formación de pontes de hidróxeno entre os átomos que forman o enlace peptídico, é dicir, un tipo de enlace non covalente, sen facer referencia á cadea lateral. Existen diferentes tipos de estrutura secundaria:
- Estrutura secundaria ordenada, (tramos repetitivos onde se encontran as hélices alfa e cadeas beta, e non repetitivos onde se encontran os xiros beta e comba beta).
- Estrutura secundaria non ordenada.
Os motivos máis comúns son a hélice alfa e a folla beta.
Hélice alfa
[editar | editar a fonte]
Os aminoácidos nunha hélice α están dispostos nunha estrutura helicoidal dextroxira, cuns 3,6 aminoácidos por volta. Cada aminoácido supón un xiro duns 100° na hélice, e os carbonos α de dous aminoácidos contiguos están separados por 1,5Å. A hélice está estreitamente empaquetada, de forma que non hai case espazo libre dentro da hélice. Todas as cadeas laterais dos aminoácidos están dispostas cara ao exterior da hélice.[5]
O grupo amino do aminoácido (n) pode establecer un enlace de hidróxeno co grupo carbonilo do 4º aminoácido seguinte (n+4). Desta forma, cada aminoácido (n) da hélice forma dúas pontes de hidróxeno cos grupos C=O e N-H do seu enlace peptídico e os do 4º aminoácido anterioer e o 4º posterior (n+4 e n-4). En total son 7 enlaces de hidróxeno por volta. Isto estabiliza enormemente a hélice.
Folla beta
[editar | editar a fonte]- Artigo principal: Folla beta.
A folla ou lámina beta fórmase polo posicionamento paralelo de dúas cadeas de aminoácidos dentro da mesma proteína, no que os grupos amino dunha das cadeas forman enlaces de hidróxeno cos grupos carboxilo da oposta. É unha estrutura moi estable que pode chegar a orixinarse por unha rotura dos enlaces de hidróxeno durante a formación da hélice alfa. As cadeas laterais desta estrutura están posicionadas sobre e baixo o plano das láminas. Ditos substituíntes non deben ser moi grandes, nin crear un impedimento estérico, xa que se vería afectada a estrutura da lámina.[6]
Estrutura terciaria
[editar | editar a fonte]- Artigo principal: Estrutura terciaria das proteínas.
É o modo en que a cadea polipeptídica se prega no espazo, é dicir, como se enrola unha determinada proteína, xa sexa globular ou fibrosa. É a disposición dos dominios no espazo.
A estrutura terciaria establécese de maneira que os aminoácidos apolares se sitúan cara ao interior e os polares cara ao exterior en medios acuosos. Isto provoca unha estabilización por interaccións hidrófobas, de forzas de van der Waals e de pontes disulfuro [7] (covalentes, entre aminoácidos de cisteína convenientemente orientados) e por medio de enlaces iónicos.
Estrutura cuaternaria
[editar | editar a fonte]- Artigo principal: Estrutura cuaternaria das proteínas.
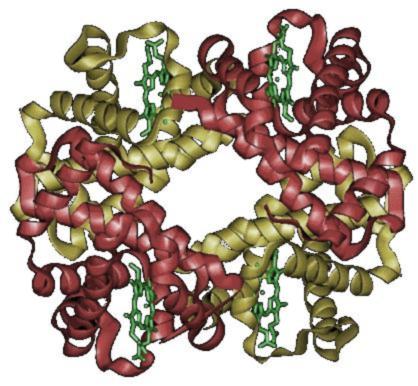
A estrutura cuaternaria deriva da conxunción de varias cadeas peptídicas que, asociadas, conforman un ente multimérico, que posúe propiedades distintas ás dos monómeros que o compoñen. Ditas subunidades asócianse entre si porr medio de interaccións non covalentes, como poden ser pontes de hidróxeno, interaccións hidrófobas ou pontes salinas. Para o caso dunha proteína constituída por dous monómeros, un dímero, este pode ser un homodímero, se os monómeros constituíntes son iguais, ou un heterodímero, se non o son.
Ángulos de rotación e representacións de Ramachandran
[editar | editar a fonte]
Nunha cadea polipeptídica defínense dous enlaces do armazón que poden rotar: un é o enlace entre o nitróxeno e o Cα, e o outro o enlace entre o Cα e o osíxeno do carbonilo. Ambos os dous definen dous ángulos de rotación:
- O ángulo de rotación φ, definido polos catro átomos sucesivos do esqueleto CO-NH-Cα-CO, que implica a dous aminoácidos.
- O ángulo de rotación ψ, definido polos catro átomos sucesivos do esqueleto: NH-Cα-CO-NH, que implica a dous aminoácidos.
A orientación do eixe de xiro considerada positiva por convención móstrase na imaxe; corresponde á das agullas do reloxo.[2]
Existen dúas excepcións nos aminoácidos que se representan nestes diagramas: a glicina, que carece dun substituínte (só ten un H), e a prolina, cíclica debido a ter unha estrutura tipo pirrol, non cumpren os requisitos requiridos para unha representación convencional [8]

Pódese describir, por tanto, a conformación do armazón de calquera residuo concreto dunha proteína especificando estes dous ángulos.[2] Con estes dous parámetros podemos describir a conformación de dito residuo nun mapa por medio dun punto, con coordenadas φ e ψ. Para determinados tipos de estrutura secundaria, como a hélice alfa, todos os residuos comparten ditos ángulos, polo que un punto no mapa en determinada posición pode describir unha estrutura secundaria. Estes mapas, denominados representacións de Ramachandran, polo bioquímico G. N. Ramachandran[9] que os usou amplamente en 1963, permiten deducir ditas conformacións.[2]
Dominios, motivos e outros elementos conformacionais
[editar | editar a fonte]- Artigo principal: Dominio proteico.
É común que algunhas zonas da proteína teñan entidade estrutural independente, e a miúdo funciós bioquímicas específicas, como, por exemplo, algunha actividade catalítica. A súa natureza depende das estruturas anteriormente citadas a todos os niveis.
A estrutura cuaternaria deriva da conxunción de varias cadeas peptídicas que, asociadas, conforman un ente multimérico con propiedades distintas.

As proteínas están organizadas en moitas unidades. Un dominio estrutural é un elemento da estrutura das proteínas que se autoestabiliza e a miúdo estabiliza os motivos conformacionais independentemente do resto da cadea da proteína. Moitos dominios son únicos e proceden dunha secuencia única dun xene ou unha familia xénica pero outros aparecen nunha variedade de proteínas. Os dominios son a miúdo seleccionados evolutivamente porque posúen unha función importante na bioloxía da proteína á que pertencen; por exemplo, "o domino de unión ao calcio da calmodulina". A enxeñaría xenética permite modificar os dominios dunha proteína para xerar proteínas quiméricas con funcións novidosas. Un motivo neste sentido refírese a unha combinación específica de elementos estruturais secundarios (como os hélice-xiro-hélice). Estes elementos chámase a miúdo superestruturas secundarias.
Adoita denominarse motivo conformacional de forma global a un tipo de motivo, como o barril beta. A estrutura dos motivos con frecuencia consta de só uns poucos elementos, por exemplo, as hélice-xiro-hélices, que só teñen tres. A “secuencia espacial” é a mesma en todas as instancias do motivo. A súa orde é bastante irregular dentro do xene subxacente. Os motivos estruturais da proteína comunmente inclúen xiros de lonxitude variable en estruturas indeterminadas, o que crea a plasticidade necesaria para unir dous elementos no espazo que non están codificados por unha secuencia de ADN adxacente nun xene. Mesmo cando están codificados os elementos estruturais secundarios dun motivo na mesma orde en dous xenes, a composición cuantitativa de aminoácidos pode variar. Isto non só é certo debido ás complicadas relacións entre a estrutura terciaria e primaria, senón por cuestións relativas ao tamaño. Aínda que na base de datos do lévedo Saccharomyces cerevisiae hai descritas unhas 6.000 proteínas,[10] hai moitos menos dominios, motivos estruturais e pregamentos. Isto é, en parte, consecuencia da evolución. Isto significa, por exemplo, que un dominio dunha proteína pode ser trasladado dunha a outra, dando así unha nova función ás proteínas. Debido a estes mecanismos, os dominios ou motivos estruturais poden ser comúns a varias familias de proteínas.
Cinética do pregamento das proteínas
[editar | editar a fonte]Aínda que o pregamento das proteínas parta dun estado liñal inicial e un final ben definidos, o proceso de pregamento non é algo brusco cun lugar de partida e un fin, senón que está pragado de intermedios temporalmente mensurables e de vital importancia. Mesmo, a estrutura final non é en absoluto estática: algúns autores imaxinan as proteínas como entidades dinámicas que continuamente mudan de estrutura, dun modo similar ao latexo cardíaco [11]
O pregamento dunha proteína é un acontecemento rápido, que se completa en apenas un segundo, topoloxicamente é un problema moi complexo. Este feito deu lugar ao paradoxo de Levinthal (de Cyrus Levinthal en 1968): un cálculo aproximado indica que unha cadea polipeptídica duns 120 residuos posúe unhas 1050 conformacións. Aínda que a molécula puidera intentar unha nova conformación cada 10−13 segundos, necesitaríanse uns 1030 anos para intentar un número significativo delas.[2] Porén, proteínas destas características préganse in vitro nun minuto.
A resolución do paradoxo pasa pola aceptación da existencia de estados de pregamento intermedios, nos que a proteína se encontra parcialmente despregada, nunha secuencia de acontecementos como a seguinte:[2]
- Proteína despregada.
- Nucleación do pregamento.
- Estados intermedios.
- Estado de glóbulo fundido.
- Reordenamentos finais.
- Proteína pregada.
Elementos moduladores
[editar | editar a fonte]Temperatura
[editar | editar a fonte]O papel da temperatura é crucial porque a enerxía cinética contida nos átomos, dálles reactividade aos aminoácidos e, por tanto, ás proteínas. Porén, existe un límite, a uns 50 °C, superado o cal as proteínas perden a súa conformación porque se desnaturalizan.
A desnaturalización, producida pola temperatura e outros axentes desnaturalizantes, ocorre a varios niveis:
- Na desnaturalización da estrutura cuaternaria, as subunidades de proteínas sepáranse ou alteran a súa posición espacial.
- A desnaturalización da estrutura terciaria implica a interrupción de:
- Enlaces covalentes entre as cadeas laterais dos aminoácidos (como as pontes disulfuro entre as cisteínas).
- Enlaces non covalentes dipolo-dipolo entre cadeas laterais polares de aminoácidos.
- Enlaces dipolo inducidos por forzas de van der Waals entre cadeas laterais non polares de aminoácidos.
- Na desnaturalización da estrutura secundaria as proteínas perden todos os patróns de repetición regulares como as hélices alfa e adoptan formas aleatorias.
- A estrutura primaria, a secuencia de aminoácidos ligados por enlaces peptídicos, non se interrompe pola desnaturalización.
pH
[editar | editar a fonte]O pH afecta ao estado iónico dos aminoácidos, que son zwitterións, que non teñen implicado o seu grupo amino nin carboxilo no enlace peptídico e, especialmente, a aqueles polares, con algún grupo cargado na súa cadea lateral. O estado de ionización deste afecta á reactividade e posibilidade, por tanto, de producir un enlace químico.[7]
Chaperonas
[editar | editar a fonte]- Artigo principal: Chaperona.
As proteínas tipo chaperona son un conxunto de proteínas presentes en todas as células cuxa función é a de axudar ao pregamento doutras proteínas, tras a súa síntese ou durante o seu ciclo de actividade (por exemplo, en defensa contra o estrés térmico).[3] Estas chaperonas non forman parte da estrutura primaria da proteína funcional, senón que só se unen a ela para axudaren no seu pregamento, ensamblaxe e transporte celular a outra parte da célula onde a proteína realiza a súa función. Os cambios de conformación tridimensional das proteínas poden estar afectados por un conxunto de varias chaperonas que traballan coordinadas, dependendo da súa propia estrutura e da dispoñibilidade das chaperonas.[12]
Existen substancias químicas non proteicas que poden estabilizar ás proteínas por medio do establecemento de pontes de hidróxeno, en substitución aos establecidos coa auga. Por exemplo, a trehalosa, un azucre que se emprega en biotecnoloxía para favorecer a viabilidade das solucións ricas en proteína mesmo en condicións de estrés ambiental [13]
As chaperonas, localizadas en todos os compartimentos celulares, agrúpanse en dúas familias xerais.[3]
- Chaperonas moleculares. Que se unen e estabilizan a proteínas despregadas ou parcialmente pregadas, evitando así que se agrupen e que sexan degradadas. Integran a familia das Hsp70 no citosol e a matriz da mitocondria, BiP no retículo endoplasmático e DnaK nas bacterias.
- Chaperoninas. Que facilitan directamente o pregamento das proteínas. Inclúen a: TriC, en eucariotas; e GroEL, en bacterias e cloroplastos.
Clasificación estrutural
[editar | editar a fonte]Desenvolvéronse moitos métodos para a clasificación estrutural das proteínas; a recompilación de datos almacénase no Protein Data Bank (PDB). Moitas bases de datos existentes clasifican as proteínas usando diferentes métodos. O SCOP, CATH e FSSP son os máis usados.
Os métodos usados poderían clasificarse en: puramente manuais, manuais e automáticos e puramente automáticos. O maior problema destes métodos é a integración dos datos. A clasificación é a mesma entre SCOP, CATH e FSSP para a maioría das proteínas que foron clasificadas, pero hai aínda algunhas diferenzas e inconsistencias.
Determinación da estrutura proteica
[editar | editar a fonte]Arredor do 90% das estruturas das proteínas dispoñibles no Protein Data Bank foron determinadas por cristalografía de raios X. Este método permite medir a densidade de distribución dos electróns da proteína nas 3 dimensións (no estado de cristalización), o que permite obter as coordenadas 3D de todos os átomos para determinar a súa posición con certeza. Aproximadamente o 9% das estruturas de proteínas coñecidas foron obtidas por técnicas de resonancia magnética nuclear, que tamén poden ser usadas para identificar estruturas secundarias.

Nótese que os aspectos das estruturas secundarias poden detectarse por medios bioquímicos como o dicroísmo circular[14] O microscopio crioelectrónico converteuse recentemente nun medio para determinar as estruturas proteicas cunha resolución baixa (menos de 5 Å ou 0,5 nm) e prevese que será unha ferramenta importante para os traballos de alta resolución na década próxima. Esta técnica é aínda un recurso importante para científicos que están traballando en complexos moi grandes de proteínas, como a cuberta e cápside dos virus e as proteínas amiloides.[15][16]
| Resolución | Interpretación |
| >4.0 | Coordenadas individuais sen significado |
| 3.0 - 4.0 | Pregamento posiblemente correcto, pero comunmente con erros. Algunhas cadeas laterais posúen mal os rotámeros. |
| 2.5 - 3.0 | Pregamento ben dilucidado salvo nalgúns pregamentos superficiais, mal modelados. Algunhas cadeas laterais longas (Lys, Glu, Gln) e outras curtas (Ser, Val, Thr) mal orientadas. |
| 2.0 - 2.5 | O número de cadeas laterais cun rotámero incorrecto é moito menor. Os erros, pequenos, son detectados normalmente. Os pregamentos superficiais están bastante ben definidos. Os ligandos e a auga son visibles. |
| 1.5 - 2.0 | Poucos residuos posúen mal o rotámero. Os erros pequenos son detectados. Os pregamentos incorrectos son moi raros, mesmo en superficie. |
| 0.5 - 1.5 | En xeral, todo está correctamente resolto. As librarías de rotámeros e os estudos xeométricos fanse a este nivel de precisión. |
Investigación
[editar | editar a fonte]Existen multitude de grupos que investigan todo o relacionado coa determinación da estrutura proteica, a súa termodinámica, cinética e implicacións. Estas últimas son de especial relevancia en neuropatoloxía, porque doenzas dexenerativas como o Alzheimer[17] ou infecciosas como a enfermidade de Creutzfeldt-Jakob [18] teñen a súa causa en alteracións na estrutura das proteínas.
As ferramentas de investigación son, en moitos casos, simulacións de pregamentos feitas con programas informáticos. Os proxectos de maior actividade en canto a computación distribuída son:
- Docking@home (Universidade de Texas)
- Folding@homeArquivado 08 de setembro de 2012 en Wayback Machine. (Universidade de Stanford)
- Proteins@home (Escola Politécnica de Francia)
- Rosetta@home (Universidade de Washington).
Notas
[editar | editar a fonte]- ↑ Pickett, S.D.; Sternberg, M.J. (1993). "Empirical scale of side-chain conformational entropy in protein folding" (w). J Mol Biol 231 (3): 825–39. doi:10.1006/jmbi.1993.1329.
- ↑ 2,00 2,01 2,02 2,03 2,04 2,05 2,06 2,07 2,08 2,09 2,10 Mathews, C. K.; Van Holde, K.E; Ahern, K.G (2003). "6". Bioquímica (3ª ed.). pp. 204 y ss. ISBN 84-7892-053-2.
- ↑ 3,0 3,1 3,2 3,3 Lodish; et al. (2005). Buenos Aires: Médica Panamericana, ed. Biología celular y molecular. ISBN 950-06-1974-3.
- ↑ Sela M, Lifson S. (1959). "The reformation of disulfide bridges in proteins". Biochim Biophys Acta 36 (2): 471–8. PMID 14444674. doi:10.1016/0006-3002(59)90188-X. PMID 14444674
- ↑ Neurath, H (1940). "Intramolecular folding of polypeptide chains in relation to protein structure". Journal of Physical Chemistry 44: 296–305. doi:10.1021/j150399a003.
- ↑ Voet, Donald ; Voet, Judith G. (2004). Wiley - Hoboken, NJ, ed. Biochemistry (3rd ed.). pp. 227–231. ISBN 047119350X.
- ↑ 7,0 7,1 Lehninger, Albert (1993). Principles of Biochemistry, 2nd Ed. Worth Publishers. ISBN 0-87901-711-2.
- ↑ Carl-Ivar Brändén, John Tooze (trad. Bernard Lubochinsky, préf. Joël Janin), Introduction à la structure des protéines, De Boeck Université, Bruxelles, 1996 ISBN 2-8041-2109-7
- ↑ Ramachandran, G.N., Sasisekharan, V. & Ramakrishnan, C. (1963) J. Mol. Biol. 7, 95–99 (1963
- ↑ Payne, W.E.; Garrels, J.I. (1997). "Yeast Protein database (YPD): a database for the complete proteome of Saccharomyces cerevisiae". Nucleic Acids Research 25 (1): 57–62. PMID 9016505. doi:10.1093/nar/25.1.57. PMID 9016505
- ↑ Alberts; et al. (2004). Barcelona: Omega, ed. Biología molecular de la célula. ISBN 54-282-1351-8.
- ↑ Ellis RJ, van der Vies SM (1991). "Molecular chaperones". Annu. Rev. Biochem. 60: 321–47. PMID 1679318. doi:10.1146/annurev.bi.60.070191.001541. PMID 1679318
- ↑ Prescott, L.M. (199). Microbiología. McGraw-Hill Interamericana de España, S.A.U. ISBN 84-486-0261-7.
- ↑ Juan Antonio Lugo-Ríos. "Dicroísmo circular" (en castelán). Arquivado dende o orixinal o 27 de outubro de 2007. Consultado o 8 de outubro de 2007.
- ↑ Chen, Shaoxia; Roseman, Alan M.; Hunter, Allison S.; Wood, Stephen P.; Burston, Steven G.; Ranson, Neil A.; Clarke, Anthony R.; Saibil, Helen R. (1994). "Location of a folding protein and shape changes in GroEL - GroES complexes imaged by cryo-electron microscopy" (w). Nature 371 (6494): 261–264. doi:10.1038/371261a0.
- ↑ Adrian, Marc; Dubochet, Jacques; Lepault, Jean; McDowall, Alasdair W. (1984). "Cryo-electron microscopy of viruses". Nature 308 (5954): 32–36. doi:10.1038/308032a0.
- ↑ Hashimoto M, Rockenstein E, Crews L, Masliah E (2003). "Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases.". Neuromolecular Med 4 (1-2): 21–36. PMID 14528050. PMID 14528050
- ↑ Baker & Ridley (1996). Humana Press, ed. Prion Disease. New Jersey. 0-89603-342-2.