Schutzgruppe


Eine Schutzgruppe (englisch protecting group – daher häufig als allgemeine Abkürzung in Formelschemata PG) ist in der Chemie ein Substituent, der während einer komplizierteren, mehrstufigen chemischen Synthese in ein Molekül eingeführt wird, um eine bestimmte funktionelle Gruppe vorübergehend zu schützen und so eine unerwünschte Reaktion an dieser Gruppe zu verhindern. Nach der Durchführung der gewünschten Reaktion an anderer Stelle des Moleküls wird die Schutzgruppe wieder abgespalten. Für viele funktionelle Gruppen sind mehrere mögliche Schutzgruppen bekannt, die sich in ihrer Stabilität und den Bedingungen für ihre Abspaltung unterscheiden.
Bei der Synthese von speziellen Verbindungsklassen mit sich wiederholenden funktionellen Gruppen – in der Regel sind dies Biomoleküle wie Peptide, Oligosaccharide oder Nukleotide – haben sich Standardsätze an Schutzgruppen etabliert. Schutzgruppen sind heute ein wichtiges Werkzeug in der Synthese von komplexen Verbindungen geworden.
Die Anforderungen an eine Schutzgruppe sind recht hoch. Dazu gehört, dass sie sich mit sehr guter Ausbeute und spezifisch an eine funktionelle Gruppe einführen lassen und ebenso unter milden Bedingungen wieder abzuspalten sein muss. Für beide Schritte sollten die Reaktionsbedingungen standardisierbar sein. Zudem muss die Schutzgruppe unter möglichst vielen Reaktionsbedingungen stabil sein. Nach Möglichkeit sollten die entstehenden Reaktionsprodukte leicht abtrennbar sein, und optimalerweise ist das Schutzgruppen-Reagenz auch noch preiswert. Je breiter der Erfahrungsschatz mit einer Schutzgruppe ist, umso besser ist die Vorhersagbarkeit der Reaktivität der Schutzgruppe.
Geschichte
[Bearbeiten | Quelltext bearbeiten]

Die Geschichte der Schutzgruppentechnik ist untrennbar verbunden mit der gezielten Verwendung verschiedener Ausgangsverbindungen für die Synthese eines Zielmoleküls. Die frühen Schutzgruppen beruhten in der Regel darauf, dass die Ausgangsverbindung so gewählt wurde, dass eine reaktive funktionelle Gruppe durch einen Rest blockiert und somit unreaktiv war. So wurden z. B. Anisole anstatt Phenole gewählt oder Ester anstelle von freien Hydroxygruppen. Erst mit der ab Anfang des 20. Jahrhunderts aufkommenden gezielten Synthese von immer komplexer werdenden Verbindungen wurde die Schutzgruppentechnik wirklich bedeutsam. Etwa ab 1960 wurde begonnen, in die Chemie der Schutzgruppen erheblichen Forschungsaufwand zu investieren. Während dieser Zeit begannen Chemiker immer komplexere Naturstoffe zu synthetisieren. Hervorzuheben sind vor allem die damaligen Arbeiten der Nobelpreisträger Robert B. Woodward, Elias J. Corey und Albert Eschenmoser, die bei der Synthese von komplexen Naturstoffen Pionierarbeit geleistet haben.[1][2]
Heute gibt es eine Vielzahl von Schutzgruppen, die in Monographien bezüglich ihrer Eigenschaften zusammengefasst sind.[3][4] Dabei gibt es neben etablierten Schutzgruppen sehr viele exotische Schutzgruppen, die nur für eine Synthese oder ein recht spezielles Gebiet entwickelt wurden.
Anforderungen an eine Schutzgruppe
[Bearbeiten | Quelltext bearbeiten]Das Einfügen und Entfernen von Schutzgruppen stellen keine produktiven Reaktionen in einer Abfolge von Syntheseschritten dar, ihr Produkt kommt dem angestrebten Endprodukt der Synthese nicht näher. Deswegen werden an Schutzgruppenreaktionen oft hohe Anforderungen bezüglich Preis, Ausbeute und Entwicklungsaufwand für die Reaktion gestellt.
Als Grundanforderungen für eine gute Schutzgruppe haben sich folgende Merkmale herausgebildet:
- Das Reagenz muss käuflich und preiswert oder leicht herstellbar sein
- Die Schutzgruppe muss einfach, spezifisch und in hohen Ausbeuten einführbar sein
- Sie muss stabil gegenüber einer möglichst großen Anzahl an Reaktionsbedingungen und Aufarbeitungs- und Reinigungsmethoden sein
- Sie muss spezifisch, hoch selektiv und in hohen Ausbeuten abspaltbar sein. Dabei sollten die Bedingungen standardisierbar sein.
- Sie darf kein neues Stereozentrum und auch kein diastereotopes Zentrum bilden
- Sie sollte einfach in NMR-Spektren erkennbar sein und möglichst wenig durch Signalüberlappung stören
Einen sehr wichtigen Aspekt stellt die hohe Selektivität der Abspaltung dar, denn es müssen häufig unterschiedliche funktionelle Gruppen unabhängig voneinander geschützt und entschützt werden. Im Idealfall ist immer nur eine von vielen Schutzgruppen vom Abspaltungsprozess betroffen. Das Verhalten von Schutzgruppen in der Praxis lässt sich, besonders wenn mehrere verschiedene Schutzgruppen in einem Molekül verwendet werden, nicht immer anhand der Literatur korrekt vorhersagen. Daher muss in manchen Fällen trotz großem Erfahrungsschatz sowohl für das Einführen als auch für das Abspalten noch erhebliche Entwicklungsarbeit während einer Synthese geleistet werden.[5][6]
Orthogonalität von Schutzgruppen
[Bearbeiten | Quelltext bearbeiten]
Orthogonalität von Schutzgruppen bedeutet, dass sich bei Verwendung mehrerer Schutzgruppen verschiedenen Typs jede Schutzgruppe einzeln und in einer beliebigen Reihenfolge aufgrund der verschiedenen Abspaltreagenzien abspalten lässt, ohne dass eine der anderen Schutzgruppen angegriffen wird. Im gezeigten Beispiel der geschützten Aminosäure Tyrosin kann der Benzylester hydrogenolytisch, die Fluorenylmethylenoxy-Gruppe (Fmoc) durch Basen (wie z. B. Piperidin) und der phenolische tert-Butylether mit Säuren (z. B. mit Trifluoressigsäure) gespalten werden.
Ein weit verbreitetes Beispiel für diese Anwendung ist die Fmoc-Peptidsynthese, die sowohl in Lösung als auch auf fester Phase eine große Bedeutung erlangt hat.[7] Die Schutzgruppen in der Festphasensynthese müssen bezüglich der Reaktionsbedingungen wie Reaktionszeit, Temperatur und Reagenzien so standardisiert sein, dass sie von einem Automaten durchgeführt werden und dabei Ausbeuten von weit über 99 % erreicht werden können, da sonst die Trennung des resultierenden Gemisches der Reaktionsprodukte praktisch unmöglich ist.[8]
-
Schematische Darstellung einer Festphasen-Peptidsynthese mit orthogonalen Schutzgruppen X und Y
-
Fmoc-Festphasen Peptidsynthese mit orthogonalen Schutzgruppen



Eine weitere wichtige Anwendung von orthogonalen Schutzgruppen ist die Kohlenhydrat-Chemie. Da Kohlenhydrate über Hydroxygruppen mit sehr ähnlicher Reaktivität verfügen, muss für eine gezielte synthetische Umsetzung der Schutz bzw. das Entschützen von einzelnen Hydroxygruppen möglich sein. Einen ähnlichen Fall stellt die Synthese von Nukleotiden dar. Hier hat man zum einen das Problem (wie bei der Peptidsynthese), dass es sich um vektorielle Moleküle handelt. Zum anderen hat man hier auch das Problem der Kohlenhydratchemie mit dem Zuckerrest der Ribose bei der Synthese von RNA-Molekülen.
Aber auch in der Synthese von komplexen Naturstoffen oder Wirkstoffen mit vielen funktionellen Gruppen ist man auf die Orthogonalität der Schutzgruppen angewiesen.[2][9]
Labilität bzw. Abspaltung von Schutzgruppen
[Bearbeiten | Quelltext bearbeiten]Bei Schutzgruppen haben sich verschiedene Reaktionsbedingungen etabliert, die dem Orthogonalitäts-Prinzip entsprechen, unter denen Schutzgruppen abgespalten werden. Man kann grob zwischen folgenden Abspaltbedingungen unterscheiden:[10]
- Säurelabile Schutzgruppen
- Basenlabile Schutzgruppen
- Fluoridlabile Schutzgruppen
- Enzymlabile Schutzgruppen
- Reduktionslabile Schutzgruppen
- Oxidationslabile Schutzgruppen
- Schutzgruppen die durch Schwermetallsalze oder deren Komplexe gespalten werden
- Photolabile Schutzgruppen
- Zwei-Stufen-Schutzgruppen
Säurelabile Schutzgruppen lassen sich durch die Einwirkung von Säuren abspalten. Die Triebkraft ist hier häufig die Bildung eines verhältnismäßig stabilen Carbokations oder ein säurekatalysiertes Gleichgewicht, das auf der Seite der freien funktionellen Gruppe liegt. Beispiele für säurelabile Schutzgruppen sind die tert-Butylester, -ether und -carbamate, welche stabile Kationen bilden, und die Acetale, bei denen in Anwesenheit von Wasser das säurekatalysierte Gleichgewicht auf der Seite der entsprechenden Aldehyde oder Ketone liegt.


Bei den basenlabilen Schutzgruppen kann man mechanistisch zwischen der basischen Hydrolyse und der baseninduzierten β-Eliminierung unterscheiden. Carbonsäureester (mit Ausnahme der tert-Butylester) werden von Hydroxidionen nukleophil angegriffen und so hydrolytisch gespalten. Amide hingegen werden selten so gespalten, da sie recht harsche Bedingungen benötigen. Eine Ausnahme stellt hier die Phthaloyl-Gruppe dar, da diese mit Hydrazin schon unter recht milden Bedingungen gespalten wird. Bei der β-Eliminierung läuft eine Reaktionskaskade ab: Zunächst wird ein Proton durch die Base abgespalten und ein Carbanion gebildet. Durch eine geeignete Abgangsgruppe wird nun die Schutzgruppe unter Bildung einer Vinylverbindung gespalten. Zum Letzteren Fall zählt vor allem die Fmoc-Gruppe.

Fluorid-Ionen bilden mit Silicium eine sehr stabile Bindung. Daher werden die Silicium-Schutzgruppen praktisch ausnahmslos durch Fluorid-Ionen gespalten. Je nach Art des Gegenions bzw. des Abspaltreagenzes können jedoch auch verschiedene Silicium-Schutzgruppen in Abhängigkeit von der sterischen Hinderung des Silicium-Atoms selektiv gespalten werden. Der Vorteil von Fluorid-labilen Schutzgruppen ist, dass keine andere Schutzgruppe unter den Abspaltbedingungen angegriffen wird.
Ester können häufig durch Enzyme wie Lipasen gespalten werden. Da Enzyme bei einem pH-Wert zwischen 5 und 9 und bei moderaten Temperaturen von etwa 30–40 °C arbeiten und zudem, was die Carbonsäure betrifft, noch sehr selektiv sind, ist diese Methode eine zwar selten benutzte, aber sehr attraktive Methode zur Schutzgruppenspaltung.

Benzylgruppen können reduktiv durch katalytische Hydrierung gespalten werden. Zum Einsatz kommen Benzylgruppen als Ether, Ester, Urethane, Carbonate oder Acetale und werden zum Schutz von Alkoholen, Carbonsäuren, Aminen und Diolen eingesetzt.
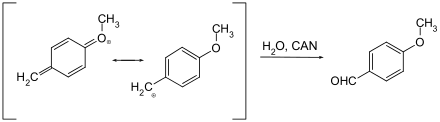
Nur wenige Schutzgruppen, die oxidativ entfernt werden können, sind gebräuchlich. Es handelt sich hier in der Regel um Methoxybenzylether. Sie können mit Cer(IV)-ammoniumnitrat (CAN) oder Dichlordicyanobenzochinon (DDQ) über ein Chinomethin gespalten werden.

Die Doppelbindung eines Allylrestes kann durch Platingruppenelemente (wie Palladium, Iridium oder Platin) zur Vinylverbindung isomerisiert werden. Die so erhaltenen Enolether bei geschützten Alkoholen oder Enamine bei geschützten Aminen können leicht sauer hydrolysiert werden.
Photolabile Schutzgruppen enthalten ein Chromophor, das durch Bestrahlung mit einer geeigneten Wellenlänge aktiviert und so abgespalten werden kann.[11] Als Beispiel sei hier die o-Nitrobenzylgruppe (ONB) aufgeführt.

Eine besondere Form von Schutzgruppen stellen die Zwei-Stufen-Schutzgruppen dar. Diese zeichnen sich durch eine hohe Stabilität aus, da die Schutzgruppe zunächst durch eine chemische Transformation in eine abspaltbare Gruppe umgewandelt werden muss. Diese Art an Schutzgruppen finden jedoch selten Anwendung, da hier ein zusätzlicher Aktivierungsschritt notwendig ist, was die Synthese um eine weitere Stufe verlängert.
Funktionelle Gruppen
[Bearbeiten | Quelltext bearbeiten]Amine
[Bearbeiten | Quelltext bearbeiten]Für die Aminofunktion ist die bei weitem größte Vielfalt an Schutzgruppen verfügbar. Dies hängt zum einen damit zusammen, dass Aminen in der Peptidsynthese eine besondere Wichtigkeit zukommt, aber auch an ihren Eigenschaften: Sie sind zum einen recht potente Nukleophile, aber auch verhältnismäßig starke Basen. Diese Eigenschaften führten dazu, dass immer neue Schutzgruppen für Amine entwickelt wurden.[12]
Viele Schutzgruppen für Amine basieren auf Carbamaten. Diese lassen sich leicht in Form von Carbonsäurechloriden einführen. Ihre Triebkraft bei der Spaltung beziehen sie durch die Bildung des sehr stabilen Kohlenstoffdioxid-Moleküls. Basierend auf unterschiedlichen Resten am Carbamat wurden verschiedene Spaltungsmöglichkeiten entwickelt. Die am häufigsten benutzten Carbamate sind die tert-Butyloxycarbonyl-, Benzyloxycarbonyl-, die Fluorenylmethylenoxycarbonyl- und die Allyloxycarbonyl-Verbindungen.
| Rest | Formel | Name | Abkürzung | Spaltung |
|---|---|---|---|---|
| tert-Butyl |  |
tert-Butyloxycarbonyl | Boc | sauer; Trifluoressigsäure (TFA) rein oder als Lösung in Dichlormethan,[13] 3 M Salzsäure in Essigsäureethylester[14] oder 10%ige Schwefelsäure in Dioxan[15] |
| Benzyl |  |
Benzyloxycarbonyl | Cbz oder Z | hydrogenolytisch; Wasserstoff und Palladium auf Aktivkohle[16], Lithium oder Natrium in flüssigem Ammoniak[17] |
| Fluorenylmethylen |  |
Fluorenylmethylenoxycarbonyl | Fmoc | basisch; 20–50 % Piperidin in Dimethylformamid (DMF)[18] oder N-Methyl-2-pyrrolidon,[19] 50 % Morpholin in DMF bei empfindlichen Glykopeptiden[20][21] |
| Allyl |  |
Allyloxycarbonyl | Alloc | übergangsmetallkatalysierte Spaltung; Metalle wie Palladium(0)– oder Nickel(0)-Komplexe[22] |
Neben den Carbamaten sind noch eine Reihe anderer N-Acyl-Derivate als Schutzgruppen von Bedeutung, aber bei weitem nicht so weit verbreitet. Dazu gehören beispielsweise die Phthalimide, die entweder durch die Umsetzung der primären Amine mit Phthalsäureanhydrid oder durch den Aufbau der Aminogruppe über eine Gabriel-Synthese zugänglich sind. Die Spaltung der Phthalimide erfolgt normalerweise durch Hydrazinhydrat oder Natriumboranat.[23] Trifluoracetamide sind überaus leicht im Basischen zu verseifen, daher dienen die durch die Umsetzung mit Trifluoressigsäureanhydrid erhaltenen Acetamide gelegentlich als Schutzgruppe für Amine.
Bei Indolen, Pyrrol und Imidazolen, also heterocyclischen Verbindungen, finden die N-Sulfonyl-Derivate als Schutzgruppe ihre Anwendung. Bei normalen Aminen ist diese Schutzgruppe häufig zu stabil. Die Darstellung erfolgt hier durch Sulfonierung mit Phenylsulfonylchlorid und dem deprotonierten Heterocyclus. Die Spaltung erfolgt durch basische Hydrolyse. N-Acyl-Derivate von primären und sekundären Aminen sind zwar relativ leicht durch die Umsetzung der Amine mit einem Arylsulfonsäurechlorid zugänglich, können aber nur schwer z. B. unter den Bedingungen einer Birch-Reduktion (Natrium in flüssigem Ammoniak) oder durch Umsetzung mit Natriumnaphthalid gespalten werden.[24]
Unter den N-Alkyl-Derivaten haben die durch Alkylierung oder reduktive Alkylierung darstellbaren N-Benzyl-Derivate eine gewisse Bedeutung. Die Spaltung erfolgt wie bei der Cbz-Gruppe reduktiv und normalerweise durch katalytische Hydrierung oder durch Birch-Reduktion. N-Alkylamine haben hier den entscheidenden Nachteil gegenüber den Carbamaten oder Amiden, dass der basische Stickstoff erhalten bleibt.
Alkohole
[Bearbeiten | Quelltext bearbeiten]Die klassische Schutzgruppe für Alkohole sind Carbonsäureester. Häufig sind die Ester von Vorstufen käuflich erhältlich oder können leicht durch Umsetzung der Alkohole mit den Säurechloriden oder Anhydriden durch eine Schotten-Baumann-Reaktion oder aber durch Umesterung erhalten werden. Die Spaltung der Ester erfolgt in der Regel durch die Umsetzung mit Nukleophilen wie den Alkalihydroxiden, Alkali–Alkoholaten oder Lithium– bzw. Magnesium–organischen Verbindungen; alternativ auch reduktiv durch Umsetzung mit komplexen Hydriden wie Lithiumaluminiumhydrid. Die Reaktivität der Ester gegenüber nukleophilen Angriffen sinkt mit der sterischen Hinderung der Carbonsäure in der Reihenfolge:
- Chloracetyl > Acetyl > Benzoyl > Pivaloyl
Die Reaktivität der Alkohole sinkt ebenfalls mit der steigenden sterischen Hinderung der Alkohole:


Die wichtigsten Ester, die als Schutzgruppen gebräuchlich sind, sind die Essigsäureester, die Benzoesäureester und die Pivalinsäureester, da diese sich nach den angegebenen Reaktivitäten differenziert voneinander abspalten lassen.
Zu den wichtigsten Schutzgruppen von Alkoholen und auch Phenolen zählen die sehr gut untersuchten und dokumentierten trisubstituierten Silylether. Dabei trägt das Silicium als organische Reste sowohl Alkyl- als auch Arylgruppen. Dieser Typ an Schutzgruppe hat den Vorteil, dass er bezüglich der Einführung und besonders bezüglich der Abspaltung sehr gut moderierbar ist. Hergestellt werden diese Ether entweder in einer Williamson-Ethersynthese aus dem Chlorsilan und einem Alkoholat-Ion oder aber durch die Verwendung von Aktivierungsreagenzien wie Imidazol.
Für rein analytische Zwecke, z. B. um ein Kohlenhydrat flüchtig zu machen und mit Hilfe von GC-MS detektieren zu können, existieren kommerziell erhältliche Reaktionskits.[25] Silylether sind grundsätzlich empfindlich gegenüber Säuren und Fluorid-Ionen. Letzteres wird meist für deren Spaltung ausgenutzt. Die kommerziellen Preise der Chlorsilane sind jedoch je nach Substitution sehr unterschiedlich. Das preiswerteste Chlorsilan ist hier das Chlortrimethylsilan (TMS-Cl), das ein Nebenprodukt der Silikon-Herstellung nach Rochow und Müller ist. Eine andere gebräuchliche Quelle der Trimethylsilyl-Gruppe ist das Hexamethyldisilazan (HMDS). Jedoch sind die Trimethylsilylether auch extrem empfindlich gegenüber sauerer Hydrolyse (beispielsweise ist schon Kieselgel als Protonendonator ausreichend) und werden daher heute selten als Schutzgruppe benutzt.
| Name | Formel | Abkürzung | Spaltung |
|---|---|---|---|
| Trimethylsilyl |  |
TMS | Kaliumfluorid, Essigsäure oder Kaliumcarbonat in Methanol[26] |
| Triethylsilyl |  |
TES | 10–100 Mal stabiler als eine TMS-Gruppe;[27] Trifluoressigsäure in Wasser/Tetrahydrofuran,[28] Essigsäure in Wasser/Tetrahydrofuran,[29] Fluorwasserstoffsäure, Pyridiniumhydrofluorid in Pyridin[30] |
| tert-Butyldimethylsilyl |  |
TBS, TBDMS | Essigsäure in Tetrahydrofuran/Wasser,[31] Pyridiniumtosylat in Methanol,[32] Trifluoressigsäure in Wasser,[33] Fluorwasserstoffsäure in Acetonitril,[34] Pyridiniumhydrofluorid in Tetrahydrofuran,[35] Tetrabutylammoniumfluorid in THF[36] |
| Triisopropylsilyl |  |
TIPS | Unter gleichen Bedingungen wie TBS aber längere Reaktionszeiten; Tetrabutylammoniumfluorid in Tetrahydrofuran, Fluorwasserstoffsäure in Acetonitril, Pyridiniumhydrofluorid in Tetrahydrofuran.[37] |
| tert-Butyldiphenylsilyl |  |
TBDPS | Unter gleichen Bedingungen wie TBS aber längere Reaktionszeiten (100–250 Mal langsamer als TBS und 5–10 Mal langsamer als TIPS); Tetrabutylammoniumfluorid in Tetrahydrofuran, Fluorwasserstoffsäure in Acetonitril, Pyridiniumhydrofluorid in Tetrahydrofuran[38] |
Eine weitere Klasse an Schutzgruppen für Alkohole sind die Alkylether. Auch hier gibt es vielfältige und orthogonale Möglichkeiten die Ether zu spalten. Aliphatische Methoxyether sind nur schwer und unter drastischen Bedingungen spaltbar, so dass diese im Allgemeinen nur bei Phenolen zum Einsatz kommen.
| Name | Formel | Abkürzung | Spaltung |
|---|---|---|---|
| Methyl | Me | In der Regel nur für Phenole gebräuchlich; Iodtrimethylsilan in Chloroform, Dichlormethan oder Acetonitril,[39] Bortribromid oder Bortrichlorid in Dichlormethan,[40] Lewis-Säuren (Aluminiumchlorid, Bortrifluorid in Gegenwart von Thiolen)[39] | |
| Benzyl |  |
Bn | reduktiv; Katalytische Hydrierung (Palladium auf Aktivkohle, Raney-Nickel oder Rhodium auf Aluminiumoxid als Katalysator)[41] |
| p-Methoxybenzyl | PMB, MPM | oxidativ; DDQ (Dichlordicyanochinon) in Dichlormethan,[42] Cerammoniumchlorid in Wasser[43] | |
| 3,4-Dimethoxbenzyl |  |
DMB, DMPM | wie PMB oxidativ; DDQ (Dichlordicyanochinon) in Dichlormethan, Cerammoniumchlorid in Wasser[44] |
| Triphenylmethyl (Trityl) |  |
Tr | sauer; Ameisensäure in Ether oder Wasser,[45] 80 % Essigsäure,[46] 1 M Salzsäure[47] |
| tert-Butyl |  |
sauer; wasserfreie Trifluoressigsäure, Bromwasserstoffsäure/Essigsäure, 4 N Salzsäure[48] | |
| Allyl | Kalium-tert-butanolat,[49] Palladium auf Aktivkohle, DABCO in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung.[50] | ||
| Allyloxycarbonyl | Alloc | Wie Allyl; Kalium-tert-butanolat, Palladium auf Aktivkohle, DABCO in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung[51] | |
| Methoxymethyl | MOM | Sauer; 6 M Salzsäure in Tetrahydrofuran/Wasser[52] | |
| Methylthiomethyl | MTM | Quecksilber(II)-chlorid/Calciumcarbonat in Acetonitril/Wasser,[53] Silbernitrat in Tetrahydrofuran/Wasser[54] | |
| (2-Methoxyethoxy)methyl | MEM | Wässrige Bromwasserstoffsäure in Tetrahydrofuran,[55] Zinkbromid in Dichlormethan[56] | |
| Benzyloxymethyl | BOM | Vergleichbar mit der Stabilität von MOM, MEM und SEM;[57] Reduktiv; Natrium in flüssigem Ammoniak,[58][59] Katalytische Hydrierung (Palladiumhydroxid auf Aktivkohle), Raney-Nickel in Ethanol[60][61] | |
| β-(Trimethylsilyl)ethoxymethyl |  |
SEM | Labiler als MEM und MOM gegenüber saurer Hydrolyse; 0.1 M Salzsäure in Methanol,[62] konzentrierte Fluorwasserstoffsäure in Acetonitril,[32] Bortrifluorid-Etherat in Dichlormethan,[63] Tetrabutylammoniumfluorid in HMPT (Hexamethylphosphorsäuretriamid) oder in Tetrahydrofuran[64][65] |
| Tetrahydropyranyl |  |
THP | Essigsäure in Tetrahydrofuran/Wasser,[66] p-Toluolsulfonsäure in Methanol[67] |
1,2-Diole
[Bearbeiten | Quelltext bearbeiten]Eine besondere Klasse von Alkoholen in der Schutzgruppen-Chemie stellen die 1,2-Diole (Glycole) dar. Die Nachbarstellung von zwei Hydroxygruppen kann man z. B. bei Zuckern dazu ausnutzen, dass man beide Hydroxygruppen abhängig voneinander als Acetal schützt. Gebräuchlich sind hier die Benzyliden–, Isopropyliden– und Cyclohexyliden– bzw. Cyclopentyliden-Acetale.

Die Herstellung der Acetale erfolgt in der Regel durch Verschieben des Gleichgewichtes eines Gemisches des Glycols mit der Carbonyl-Komponente durch Entfernen des Reaktionswassers oder durch Umacetalisierung mit einem einfachen Acetal und dem Entfernen des entstehenden Alkohols aus dem Reaktionsgemisch.

Gerade in der Zuckerchemie wird hier die unterschiedliche Stellung der Hydroxygruppen zueinander ausgenutzt, um diese in bestimmter stereochemischer Abhängigkeit selektiv zu schützen. So reagieren (neben den anderen möglichen Kombinationen) die beiden benachbarten Hydroxygruppen bevorzugt miteinander, welche die stabilste Konformation bilden.[68][69]

Acetale können grundsätzlich in wässrigen sauren Lösungsmitteln wieder gespalten werden. Einen besonderen Fall stellt hier die Benzyliden-Schutzgruppe dar, die auch reduktiv gespalten werden kann. Dies erfolgt entweder durch katalytische Hydrierung oder durch den Hydriddonor Diisobutylaluminiumhydrid (DIBAL). Die Spaltung durch DIBAL entschützt jedoch nur eine Alkoholgruppe, da der Benzylrest auf der zweiten und sterisch gehinderteren Hydroxygruppe als Benzylether verbleibt.[70][71]

Carbonylgruppen
[Bearbeiten | Quelltext bearbeiten]Carbonylgruppen sind vor allem durch nukleophile Angriffe wie Grignard-Reagenzien oder von Hydrid-Ionen gefährdet. Aldehyde können zusätzlich noch zu Carbonsäuren oxidiert werden. Aber auch unerwünschte Reaktionen, die durch säure- und basen-katalysierte Reaktionen der Carbonylgruppe wie z. B. Aldolreaktionen können durch eine geeignete Schutzgruppe verhindert werden.
Die gebräuchlichsten Schutzgruppen für Carbonylgruppen sind Acetale und hier besonders cyclische Acetale mit Diolen. Daneben werden auch cyclische Acetale mit 1,2-Hydroxythiolen oder Dithioglycolen verwendet – die sogenannten O,S– bzw. S,S-Acetale.


Für Acetale als Schutzgruppe für Carbonylverbindungen gilt grundsätzlich das gleiche wie für Acetale als Schutzgruppe für 1,2-Diole. Sowohl die Herstellung als auch die Spaltung sind naturgemäß identisch. Allerdings spielt bei Acetalen als Schutzgruppe der Prozess einer Umacetalisierung eine untergeordnete Rolle, und sie werden in der Regel aus den Glycolen durch Wasserabspaltung hergestellt. Modernere Varianten verwenden hier auch Glycole, bei welchen die Hydroxy-Wasserstoff-Atome durch eine Trimethylsilyl-Gruppe ersetzt wurden.[72][73] Normalerweise finden einfache Glycole wie das Ethylenglycol oder das 1,3-Propandiol als Diole für die Acetale Verwendung.
Acetale können unter sauren wässrigen Bedingungen gespalten werden. Dabei werden als Säuren die Mineralsäuren verwendet. Cosolvenz ist häufig Aceton, das als Lösungsvermittler benutzt wird. Als nicht-saure Abspaltmethode kann mit einem Palladium(II)-chlorid-Acetonitril-Komplex in Aceton[74] oder mit Eisen(III)-chlorid auf Kieselgel, aufgezogen in Chloroform,[75] gearbeitet werden.
Cyclische Acetale sind sehr viel stabiler gegenüber saurer Hydrolyse als acyclische Acetale. Daher werden praktisch ausschließlich acyclische Acetale benutzt, wenn eine sehr milde Abspaltung nötig ist oder wenn zwei verschiedene geschützte Carbonylgruppen bezüglich ihrer Freisetzung differenziert werden müssen.[76]
Acetale finden jedoch neben ihrer alleinigen Funktion als Schutzgruppe zusätzlich noch Anwendung als chirales Hilfsreagenz. So können Acetale von chiralen Glycolen wie z. B. Derivate der Weinsäure mit hoher Selektivität asymmetrisch geöffnet werden. Dies ermöglicht den Aufbau neuer Chiralitätszentren.[77]

Neben den O,O-Acetalen haben auch noch die S,O- und S,S-Acetale eine, wenn auch geringere, Bedeutung als Carbonylschutzgruppe. Thiole, die man zur Herstellung dieser Acetale einsetzen muss, haben einen sehr unangenehmen Geruch und sind giftig, was die Anwendung sehr einschränkt. Thioacetale und die gemischten S,O-Acetale sind, verglichen mit den reinen O,O-Acetalen, sehr viel stabiler gegenüber saurer Hydrolyse. Dies ermöglicht die selektive Spaltung dieser in Gegenwart von Schwefel-geschützten Carbonylgruppen.
Die Herstellung der S,S-Acetale erfolgt normalerweise analog der O,O-Acetale durch saure Katalyse aus den Dithiolen und der Carbonylkomponente. Aufgrund der großen Stabilität der Thioacetale liegt das Gleichgewicht auf der Seite der Acetale. Es muss zum Unterschied zu den O,O-Acetalen kein Reaktionswasser entfernt werden, um das Gleichgewicht zu verschieben.[78]
S,O-Acetale werden um den Faktor 10.000 schneller hydrolysiert als die entsprechenden S,S-Acetale. Ihre Herstellung erfolgt in Analogie zu diesen aus den Thioalkoholen. Auch ihre Spaltung erfolgt unter vergleichbaren Bedingungen und vorwiegend durch Quecksilber(II)-Verbindungen in wässrigem Acetonitril.[79]
Für Aldehyde ist eine temporäre Schützung der Carbonylgruppe in Anwesenheit von Ketonen als Halbaminal-Anion beschrieben. Hier wird ausgenutzt, dass Aldehyde eine sehr viel höhere Carbonylaktivität aufweisen als Ketone und dass viele Additionsreaktionen reversibel sind.[80][81]

Carboxygruppen
[Bearbeiten | Quelltext bearbeiten]Die wichtigsten Schutzgruppen für Carboxy-Gruppen sind die Ester von verschiedenen Alkoholen. Daneben sind auch noch Ortho-Ester und Oxazoline in Gebrauch, aber von untergeordneter Bedeutung. Für die Herstellung von Carbonsäureestern gibt es grundsätzlich verschiedene Methoden:[82]
- Direkte Veresterung von Carbonsäuren und Alkoholkomponente. Aufgrund der ungünstigen Gleichgewichtslage bei der Reaktion zwischen Alkoholen und Carbonsäuren muss das Gleichgewicht entweder durch das Entfernen des Reaktionswassers oder aber durch das Arbeiten mit großen Überschüssen an Alkohol erfolgen. Dazu muss der Alkohol jedoch sehr preiswert sein. Diese Reaktion ist säurekatalysiert (Schwefelsäure, p-Toluolsulfonsäure oder saure Ionentauscher sind die gebräuchlichsten Veresterungskatalysatoren).
- Die Reaktion von Säureanhydriden oder Säurechloriden mit Alkoholen in Gegenwart von Hilfsbasen. Als Hilfsbase findet hier häufig Pyridin, Diisopropylethylamin oder Triethylamin Anwendung. Diese Reaktion kann mit 4-N,N-Dimethylaminopyridin katalysiert werden, was die Reaktionsgeschwindigkeit im Vergleich zu reinem Pyridin um den Faktor 104 steigert. Im Vergleich zur direkten Veresterung erfolgen diese Methoden unter recht milden Bedingungen.
- Die Reaktion von Carbonsäuresalzen mit Alkylhalogeniden ist eine weitere Methode zur Herstellung von Carbonsäureestern.
- Die Reaktion von Carbonsäuren mit Isobuten ist eine sanfte Methode zur Herstellung von tert-Butylestern. Hier wird Isobuten mit der Carbonsäure in Gegenwart einer starken Säure wie Schwefelsäure umgesetzt.
- Die Reaktion von Carbonsäuren mit Diazoalkanen ist eine sehr sanfte und quantitative Methode, um Ester herzustellen. Sie wird aufgrund der schlechten Zugänglichkeit von komplexen Diazoalkanen jedoch meist nur für die Herstellung von Methyl- und Benzhydril-Estern benutzt.
Neben diesen klassischen Methoden der Veresterung wurden für spezielle Reaktionen weitere und modernere Methoden entwickelt.
- Die Aktivierung der Carbonsäure mit Dicyclohexylcarbodiimid und Umsetzung des so erhaltenen O-Acylisoharnstoffes mit der Alkoholkomponente in Gegenwart von 4-N,N-Dimethylaminopyridin (Steglich-Veresterung).
- Die Aktivierung der Carbonsäure durch Herstellung eines gemischten Anhydrides mit der 2,4,6-Trichlorbenzoesäure durch Umsetzung der Carbonsäure mit Benzoylchlorid in Gegenwart von 4-N,N-Dimethylaminopyridin und Triethylamin. Das gemischte Anhydrid wird in situ hergestellt und sofort weiter mit der Alkoholkomponente umgesetzt (Yamaguchi-Veresterung).
- Die Aktivierung der Alkoholkomponete durch die Umsetzung unter Mitsunobu-Bedingungen mit Diethylazodicarboxylat und Triphenylphosphin und anschließenden Umsetzung in situ mit der Carbonsäure (Mitsunobu-Veresterung).
Als Alkoholkomponente können verschiedene Gruppen dienen. Sehr gebräuchlich sind hier jedoch die Methyl-, die tert-Butyl, die Benzyl- und die Allylester. Darüber hinaus kommen noch eine Reihe Schutzgruppen hinzu, welche sich aus den Ether-Schutzgruppen der Hydroxygruppen herleiten. Die spezifischen Abspaltbedingungen sind jedoch häufig sehr ähnlich. Grundsätzlich kann jeder Ester in Anwesenheit von Hydroxidionen in wässrig-alkoholischer Lösung hydrolysiert werden. Bei empfindlicheren Substraten verwendet man jedoch häufig Lithiumhydroxid in Tetrahydrofuran und in Anwesenheit von Methanol. Für die Hydrolysetendenz gelten naturgemäß die gleichen Regeln wie bei den Estern als Alkoholschutzgruppe.
| Name | Formel | Abkürzung | Spaltung | Besondere Herstellung |
|---|---|---|---|---|
| Methyl | Me | nukleophil-alkalisch durch Metallhydroxide oder Carbonate in wässrigem Methanol oder Tetrahydrofuran,[83][84] Alkalimetallhalogenide in feuchten aprotischen Lösungsmitteln wie Dimethylsulfoxid, N,N-Dimethylformamid in der Hitze,[85][86][87] Enzymatisch z. B. durch Schweineleberesterase[88][89] | Diazomethan in Diethylether,[90][91][92] Caesiumcarbonat und Methyliodid in N,N-Dimethylformamid,[93] Methanol und katalytisch Trimethylsilylchlorid[94] | |
| tert-Butyl | |
tert-Bu | sauer; Trifluoressigsäure (rein oder in Dichlormethan),[95] Ameisensäure, p-Toluolsulfonsäure[96] | Isobuten in Dioxan und katalytisch Schwefelsäure[97][98][99] |
| Benzyl | |
Bn | hydrogenolytisch; Wasserstoff/Palladium auf Aktivkohle[100] | |
| Benzhydryl |  |
hydrogenolytisch; Wasserstoff/Palladium auf Aktivkohle (sehr leicht zu spalten)[101] | ||
| Allyl | Allyl | analog der Ether; Kalium-tert-Butanolat, Palladium auf Aktivkohle, DABCO (1,4-Diazabicyclo[2.2.2]octan) in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung[102] |
Alkene
[Bearbeiten | Quelltext bearbeiten]Alkene werden und müssen selten durch eine Schutzgruppe geschützt werden. Sie sind in der Regel nur durch elektrophile Angriffe, Isomerisierung und bei der katalytischen Hydrierung von unerwünschten Nebenreaktionen betroffen. Grundsätzlich kennt man für Alkene zwei Schutzgruppen:
- Die temporäre Halogenierung mit Brom zur trans-1,2-Dibromalkyl-Verbindung: Die Regeneration des Alkens erfolgt unter Wiederherstellung der Konformation durch elementares Zink[103][104][105][106][107] oder mit Titanocendichlorid.[108]
- Die Schützung durch eine Diels-Alder-Reaktion: Die Umsetzung eines Alkens mit einem Dien führt zu einem cyclischen Alken, welches jedoch durch elektrophile Angriffe ähnlich gefährdet ist wie das ursprüngliche Alken. Die Abspaltung des als Schutzgruppe dienenden Diens erfolgt thermisch, da es sich bei einer Diels-Alder-Reaktion um eine reversible bzw. Gleichgewichtsreaktion handelt.[109][110][111]

Alkine
[Bearbeiten | Quelltext bearbeiten]Für Alkine kennt man ebenfalls zwei Typen von Schutzgruppen. Bei terminalen Alkinen ist es manchmal notwendig, das acide Wasserstoffatom zu maskieren. Dies erfolgt normalerweise durch Deprotonierung (mittels starker Basen wie Methylmagnesiumbromid oder Butyllithium in Tetrahydrofuran/Dimethylsulfoxid) und anschließender Umsetzung mit Chlortrimethylsilan zum terminal TMS-geschützten Alkin. Die Abspaltung erfolgt hydrolytisch – mit Kaliumcarbonat in Methanol – oder durch Fluorid-Ionen wie beispielsweise mittels Tetrabutylammoniumfluorid.[112]

Um die Dreifachbindung selbst zu schützen, wird manchmal ein Komplex der Alkin-Verbindung mit Dicobaltoctacarbonyl benutzt. Die Abspaltung des Cobalts erfolgt durch Oxidation.[113][114][115][116][117]

Anwendungen
[Bearbeiten | Quelltext bearbeiten]Schutzgruppen finden in weiten Bereichen der organischen Synthesechemie ihre Anwendung. Dies betrifft sowohl die Laborsynthesen als auch großtechnische Synthesen von komplexen Wirkstoffen. Sobald eine funktionelle Gruppe sich als störend erweist oder aber unerwünscht angegriffen werden kann, findet die Schutzgruppentechnik ihre Anwendung. Beinahe bei jeder Synthese eines komplexen Zielmoleküls kommen Schutzgruppen zum Einsatz.[1][2] Da sowohl das Einführen als auch das Abspalten der Schutzgruppen neben dem Aufwand auch einen Ausbeuteverlust zur Folge hat, ist es erstrebenswert, ohne Schutzgruppen auszukommen, was jedoch oft nur schwer zu realisieren ist.
In der automatisierten Synthese von Peptiden und Nukleotiden ist die Schutzgruppenchemie ein integraler Bestandteil des Synthesekonzepts.[7] Aus der Zuckerchemie sind Schutzgruppen aufgrund der sehr ähnlichen Hydroxygruppen in den Zuckermolekülen ebenfalls nicht wegzudenken.[68]
Ein wichtiges Beispiel für die industrielle Anwendung der Schutzgruppentechnik ist die Synthese von Ascorbinsäure (Vitamin C) nach Reichstein.

Um eine Oxidation der sekundären Alkohole durch Kaliumpermanganat zu verhindern, werden diese durch Acetalisierung mit Aceton geschützt und nach der Oxidation der primären Hydroxygruppe zur Carbonsäure wieder entschützt.[118]
Ein sehr spektakuläres Beispiel aus der Naturstoffsynthese zur Anwendung von Schutzgruppen ist die Totalsynthese von Palytoxin-Carbonsäure durch die Arbeitsgruppe um Yoshito Kishi aus dem Jahre 1994.[119] Hier mussten 42 Funktionelle Gruppen (39 Hydroxygruppen, ein Diol, eine Aminogruppe und eine Carbonsäuregruppe) geschützt werden. Dies erfolgte durch acht verschiedene Schutzgruppen (ein Methylester, fünf Acetat-Gruppen, 20 TBDMS-Ether, neun p-Methoxybenzylether, vier Benzoate, ein Methyl-halbacetal, ein Acetal mit Aceton und ein SEM-Ester).[120]

Das Einführen oder Verändern einer Schutzgruppe beeinflusst bisweilen auch die Reaktivität des Gesamtmoleküls. Als Beispiel sei hier ein Ausschnitt aus der Synthese eines Analogons vom Mitomycin C von Danishefsky gezeigt.[121]

Der Wechsel der Schutzgruppe von einem Methylether zu einem MOM-Ether verhindert hier das Öffnen des Epoxides zum Aldehyd.
Eine bedeutende Anwendung der Schutzgruppenchemie findet sich in der automatisierten Synthese von Peptiden und Nucleosiden. Bei der Peptidsynthese durch automatische Synthesizer wird die Orthogonalität der Fmoc-Gruppe (basische Spaltung), der tert-Butyl-Gruppe (saure Spaltung) und diverse Schutzgruppen für funktionelle Gruppen in der Seitenkette der Aminosäuren ausgenutzt.[7] Bei der automatisierten Nukleotid-Synthese von DNA- und RNA-Sequenzen nach der Phosphoramidit-Synthese werden bis zu vier unterschiedliche Schutzgruppen pro Baustein verwendet. Hier erfolgt sogar Redox-Chemie an den geschützten Phosphor-Atomen. Der aufgrund der höheren Reaktivität eingesetzte dreiwertige Phosphor ist mit einer Cyanoethyl-Schutzgruppe am freien Sauerstoff versehen. Nach dem Kupplungsschritt folgt die Oxidation zum Phosphat, wobei die Schutzgruppe erhalten bleibt. Freie OH-Gruppen, die im Kopplungsschritt nicht reagiert haben, werden in einem Zwischenschritt acetyliert. Diese zusätzlich eingeführte Schutzgruppe verhindert dann, dass diese OH-Gruppe in den nächsten Zyklen koppeln kann.[122]

In der Regel ist das Einführen einer Schutzgruppe unproblematisch. Die Schwierigkeiten liegen eher in ihrer Stabilität und beim selektiven Abspalten. Auftretende Probleme bei Synthesestrategien mit Schutzgruppen werden nur selten in der Fachliteratur dokumentiert.[123]
Atomökonomie
[Bearbeiten | Quelltext bearbeiten]Synthesen unter Einsatz von Schutzgruppen weisen in der Regel eine geringe Atomökonomie auf.[124][125][126] Manchmal muss der Umweg der Verwendung von Schutzgruppen eingeschlagen werden, um unerwünschte Konkurrenzreaktionen auszuschalten und die angestrebte Selektivität einer Synthese zu erreichen.[127] Bei der Synthese komplexer Strukturen sind Schutzgruppenstrategien oft unverzichtbar.
Als Beispiel für eine Schutzgruppenstrategie im Vergleich zu einer schutzgruppenfreien Synthese seien die Synthesen von Hapalindol U gegenübergestellt. Während die Synthese von Hideaki Muratake aus dem Jahr 1990 Tosyl als Schutzgruppe verwendet,[128][129][130] wurde in der Synthese von Phil S. Baran aus dem Jahre 2007 auf jede Schutzgruppe verzichtet.[131] Dabei wurde die Zahl der Syntheseschritte maßgeblich verringert.
-
Hapalindol U Muratake 1990 Ts-Schutzgruppensynthese (Schutzgruppen in blau.)
-
Hapalindol U Baran 2007 Schutzgruppenfreie Synthese


Literatur
[Bearbeiten | Quelltext bearbeiten]- Philip J. Kocieński: Protecting Groups, 1. Auflage, Georg Thieme Verlag, Stuttgart 1994, ISBN 3-13-135601-4.
- Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, 4th Ed., John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0.
- Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2192–2219; doi:10.1002/ange.19961081805.
- Krzysztof Jarowicki, Philip Kocieński: „Protecting groups“, in: J. Chem. Soc., Perkin Trans. 1, 1998, S. 4005–4037; doi:10.1039/A803688H.
Weblinks
[Bearbeiten | Quelltext bearbeiten]- Universität Marburg: Schutzgruppen in der organischen Synthesechemie (PDF; 284 kB)
- Organic-Reaction.com: Protecting Group
- Organic-Chemistry.org: Protecting Groups
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b Kyriacos C. Nicolaou, Erik J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, 1996, ISBN 3-527-29284-5.
- ↑ a b c Kyriacos C. Nicolaou, Scott A. Snyder: Classics in Total Synthesis II, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003, ISBN 3-527-30684-6.
- ↑ Philipp J. Kocieński: Protecting Groups, 1. Auflage, Georg Thieme Verlag, Stuttgart 1994, ISBN 3-13-135601-4.
- ↑ Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, Fourth Ed. John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0.
- ↑ P.J. Kocieński: Protecting Groups, S. 245–250.
- ↑ Dietrich Spitzner, Kai Oesterreich: „Anionically Induced Domino Reactions – Synthesis of a Norpatchoulenol-Type Terpene“, in: European Journal of Organic Chemistry, 2001, 10; S. 1883–1886; doi:10.1002/1099-0690(200105)2001:10<1883::AID-EJOC1883>3.0.CO;2-M.
- ↑ a b c Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis. Reprint 2004, Oxford University Press, ISBN 0-19-963724-5.
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis, S. 10–12.
- ↑ Kyriacos C. Nicolaou, Eric J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, VCH Verlagsgesellschaft mbH, Weinheim, 1996, S. 711–729, ISBN 3-527-29284-5.
- ↑ Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2195–2200; doi:10.1002/ange.19961081805.
- ↑ V.N. Rajasekharan Pillai: „Photoremovable Protecting Groups in Organic Synthesis“, in: Synthesis, 1980, S. 1–26.
- ↑ P.J. Kocieński: Protecting Groups, S. 186.
- ↑ Naomi Sakai, Yasufumi Ohfune: „Total synthesis of galantin I. Acid-catalyzed cyclization of galantinic acid“, in: J. Am. Chem. Soc., 1992, 114, S. 998–1010; doi:10.1021/ja00029a031.
- ↑ Glenn L. Stahl, Roderich Walter, Clarck W. Smith: „General procedure for the synthesis of mono-N-acylated 1,6-diaminohexanes“, in: J. Org. Chem., 1978, 43, S. 2285–2286; doi:10.1021/jo00405a045.
- ↑ R.A. Houghton, A. Beckman, J.M. Ostresh: Int. J. Pept. Protein Res., 1986, 27, S. 653.
- ↑ P.J. Kocieński: Protecting Groups, S. 195.
- ↑ Robert M. Williams, Peter J. Sinclair, Dongguan Zhai, Daimo Chen: „Practical asymmetric syntheses of α-amino acids through carbon-carbon bond constructions on electrophilic glycine templates“, in: J. Am. Chem. Soc., 1988, 110, S. 1547–1557; doi:10.1021/ja00213a031.
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis, S. 27–30.
- ↑ Gregg B. Fields: Methods for Removing the Fmoc Group. (PDF; 663 kB) In: Michael W. Pennington, Ben M. Dunn (Hrsg.): Peptide Synthesis Protocols Band 35, 1995, ISBN 978-0-89603-273-6, S. 17–27.
- ↑ B. Liebe, H. Kunz: Festphasensynthese eines tumorassoziierten Sialyl-Tn-Antigen-Glycopeptids mit einer Partialsequenz aus dem “Tandem Repeat” des MUC-1-Mucins In: Angew. Chem. Band 109, 1997, S. 629–631.
- ↑ ChemPep Inc.: Fmoc Solid Phase Peptide Synthesis. Abgerufen am 16. November 2013.
- ↑ P.J. Kocieński: Protecting Groups, S. 199–201.
- ↑ John O. Osby, Michael G. Martin, Bruce Ganem: An Exceptionally Mild Deprotection of Phthalimides, in: Tetrahedron Lett., 1984, 25, S. 2093–2096; doi:10.1016/S0040-4039(01)81169-2.
- ↑ P.J. Kocieński: Protecting Groups, S. 220–227.
- ↑ P. Vouros: „Chemical derivatization in gas chromatographie-mass spectrometrie“, in: „Mass Spectrometrie“, Degger, New York, 1979, Bd. 2, S. 129.
- ↑ P.J. Kocieński: Protecting Groups, S. 29.
- ↑ P.J. Kocieński: Protecting Groups, S. 31.
- ↑ Tod K Jones, Robert A. Reamer, Richard Desmond, Sander G. Mills: „Chemistry of tricarbonyl hemiketals and application of Evans technology to the total synthesis of the immunosuppressant (−)-FK-506“, in: J. Am. Chem. Soc., 1990, 112, S. 2998–3017; doi:10.1021/ja00164a023.
- ↑ Dieter Seebach, Hak-Fun Chow, Richard F.W. Jackson, Marius A. Sutter, Suvit Thaisrivongs, Jürg Zimmermann: „(+)-11,11′-Di-O-methylelaiophylidene – preparation from elaiophylin and total synthesis from (R)-3-hydroxybutyrate and (S)-malate“, in: Liebigs Ann. Chem., 1986, S. 1281–1308; doi:10.1002/jlac.198619860714.
- ↑ David A. Evans, Stephen W. Kaldor, Todd K. Jones, Jon Clardy, Thomas J. Stout: „Total synthesis of the macrolide antibiotic cytovaricin“, in: J. Am. Chem. Soc., 1990, 112, S. 7001–7031; doi:10.1021/ja00175a038.
- ↑ James A. Marshall, Richard Sedrani: „A convergent, highly stereoselective synthesis of a C-11-C-21 subunit of the macbecins“, in: J. Org. Chem., 1991, 56, S. 5496–5498; doi:10.1021/jo00019a004.
- ↑ a b James D. White, Motoji Kawasaki: „Total synthesis of (+)-latrunculin A“, in: J. Am. Chem. Soc., 1990, 112, S. 4991–4993; doi:10.1021/ja00168a071.
- ↑ Morris J. Robins, Vicente Samano, Mark D. Johnson: „Nucleic acid-related compounds. 58. Periodinane oxidation, selective primary deprotection, and remarkably stereoselective reduction of tert-butyldimethylsilyl-protected ribonucleosides. Synthesis of 9-(β-D-xylofuranosyl)adenine or 3'-deuterioadenosine from adenosine“, in: J. Org. Chem., 1990, 55, S. 410–412; doi:10.1021/jo00289a004.
- ↑ R. Roger F. Newton, Derek P. Reynolds, Colin F. Webb, Stanley M. Roberts: „A short and efficient total synthesis of (±) prostaglandin D2 methyl ester involving a new method for the cleavage of a dimethyl-t-butylsilyl ether“, in: J. Chem. Soc., Perkin Trans. 1, 1981, S. 2055–2058; doi:10.1039/P19810002055.
- ↑ Kyriacos C. Nicolaou, R. A. Daines, T. K. Chakraborty: „Total synthesis of amphoteronolide B“, in: J. Am. Chem. Soc., 1987, 109, S. 2208–2210; doi:10.1021/ja00241a063.
- ↑ Leo A. Paquette, Annette M. Doherty, Christopher M. Rayner: „Total synthesis of furanocembranolides. 1. Stereocontrolled preparation of key heterocyclic building blocks and assembly of a complete seco-pseudopterane framework“, in: J. Am. Chem. Soc., 1991, 109, S. 3910–3926; doi:10.1021/ja00036a045.
- ↑ P.J. Kocieński: Protecting Groups, S. 40.
- ↑ P.J. Kocieński: Protecting Groups, S. 38–39.
- ↑ a b P.J. Kocieński: Protecting Groups, S. 43.
- ↑ J. F. W. McOmie, D. E. West: 3,3′-Dihyroxybiphenyl In: Organic Syntheses. 49, 1969, S. 50, doi:10.15227/orgsyn.049.0050; Coll. Vol. 5, 1973, S. 412 (PDF).
- ↑ P.J. Kocieński: Protecting Groups, S. 46–49.
- ↑ Yuji Oikawa, Tadao Yoshioka, Osamu Yonemitsu: „Specific removal of o-methoxybenzyl protection by DDQ oxidation“, in: Tetrahedron Lett., 1982, 23, S. 885–888; doi:10.1016/S0040-4039(00)86974-9.
- ↑ Rolf Johansson, Bertil Samuelsson: „Regioselective reductive ring-opening of 4-methoxybenzylidene acetals of hexopyranosides. Access to a novel protecting-group strategy. Part 1“, in: J. Chem. Soc., Perkin Trans. 1, 1984, S. 2371–2374; doi:10.1039/P19840002371.
- ↑ Literatur wie p-Methoxybenzyl.
- ↑ Michel Bessodes, Dimitri Komiotis, Kostas Antonakis: „Rapid and selective detritylation of primary alcohols using formic acid“, in: Tetrahedron Lett., 1986, 27, S. 579–580; doi:10.1016/S0040-4039(00)84045-9.
- ↑ B. Helferich: Carbonhydr. Chem. Biochem., 1948, 3, S. 79.
- ↑ M.L. García, J. Pascual, L. Borràs, J.A. Andreu, E. Fos, D. Mauleón, G. Carganico, F. Arcamone: „Synthesis of new ether glycerophospholipids structurally related to modulator“, in: Tetrahedron, 1991, 47, S. 10023–10034; doi:10.1016/S0040-4020(01)96051-X.
- ↑ P.J. Kocieński: Protecting Groups, S. 59–60.
- ↑ P.J. Kocieński: Protecting Groups, S. 62.
- ↑ R.E. Ireland, D.W. Norbeck: „Convergent synthesis of polyether ionophore antibiotics: the synthesis of the monensin bis(tetrahydrofuran) via the Claisen rearrangement of an ester enolate with a β-leaving group“, in: J. Am. Chem. Soc., 1985, 107, S. 3279–3285; doi:10.1021/ja00297a038.
- ↑ Literatur siehe Allyl.
- ↑ Paul A. Wender, Carlos R. D. Correia: „Intramolecular photoinduced diene-diene cyaloadditions: a selective method for the synthesis of complex eight-membered rings and polyquinanes“, in: J. Am. Chem. Soc., 1987, 109, S. 2523–2525; doi:10.1021/ja00242a053.
- ↑ Elias J. Corey, Mark G. Bock: „Protection of primary hydroxyl groups as methylthiomethyl ethers“, in: Tetrahedron Lett., 1975, 16, S. 3269–3270; doi:10.1016/S0040-4039(00)91422-9.
- ↑ Elias J. Corey, Duy H. Hua, Bai Chuan Pan, Steven P. Seitz: „Total synthesis of aplasmomycin“, in: J. Am. Chem. Soc., 1982, 104, S. 6818–6820; doi:10.1021/ja00388a074.
- ↑ Serge David, Annie Thieffry, Alain Veyrières: „A mild procedure for the regiospecific benzylation and allylation of polyhydroxy-compounds via their stannylene derivatives in non-polar solvents“, in: J. Chem. Soc., Perkin Trans. 1, 1981, S. 1796–1801; doi:10.1039/P19810001796.
- ↑ Kaoru Fuji, Shigetoshi Nakano, Eiichi Fujita: „An Improved Method for Methoxymethylation of Alcohols under Mild Acidic Conditions“, in: Synthesis, 1975, S. 276–277.
- ↑ P.J. Kocieński: Protecting Groups, S. 77.
- ↑ H. Nagaoka, W. Rutsch, G. Schmidt, H. Ito, M.R. Johnson, Y. Kishi: „Total synthesis of rifamycins. 1. Stereocontrolled synthesis of the aliphatic building block“, in: J. Am. Chem. Soc., 1980, 102, S. 7962–7965; doi:10.1021/ja00547a037.
- ↑ W. Clark Still, Dominick Mobilio: „Synthesis of asperdiol“, in: J. Org. Chem., 1983, 48, S. 4785–4786; doi:10.1021/jo00172a070.
- ↑ Masahiro Hirama, Mitsuko Uei: „A chiral total synthesis of compactin“, in: J. Am. Chem. Soc., 1982, 104, S. 4251–4253; doi:10.1021/ja00379a037.
- ↑ W. Clark Still, Shizuaki Murata, Gilbert Revial, Kazuo Yoshihara: „Synthesis of the cytotoxic germacranolide eucannabinolide“, in: J. Am. Chem. Soc., 1983, 105, S. 625–627; doi:10.1021/ja00341a055.
- ↑ Robert C. Gadwood, Renee M. Lett, Jane E. Wissinger: „Total synthesis of (±)-poitediol and (±)4-epipoitediol“, in: J. Am. Chem. Soc., 1984, 106, S. 3869–3870; doi:10.1021/ja00325a032.
- ↑ Steven D. Burke, Gregory J. Pacofsky: „The ester enolate claisen rearrangement. Total synthesis of (±)-ethisolide“, in: Tetrahedron Lett., 1986, 27, S. 445–448; doi:10.1016/S0040-4039(00)85501-X.
- ↑ Toshiyuki Kan, Masaru Hashimoto, Mitsutoshi Yanagiya, Haruhisa Shirahama: „Effective deprotection of 2-(trimethylsilylethoxy)methylated alcohols (SEM ethers). Synthesis of thyrsiferyl-23 acetate“, in: Tetrahedron Lett., 1988, 29, S. 5417–5418; doi:10.1016/S0040-4039(00)82883-X.
- ↑ Joseph P. Marino, Scott L. Dax: „An efficient desilylation method for the generation of o-quinone methides: application to the synthesis of (+)- and (−)-hexahydrocannabinol“, in: J. Org. Chem., 1984, 49, S. 3671–3672; doi:10.1021/jo00193a051.
- ↑ Karel F. Bernady, M. Brawner Floyd, John F. Poletto, Martin J. Weiss: „Prostaglandins and congeners. 20. Synthesis of prostaglandins via conjugate addition of lithium trans-1-alkenyltrialkylalanate reagents. A novel reagent for conjugate 1,4-additions“, in: J. Org. Chem., 1979, 44, S. 1438–1447; doi:10.1021/jo01323a017.
- ↑ Elias J. Corey, Haruki Niwa, Jochen Knolle: „Total synthesis of (S)-12-hydroxy-5,8,14-cis,-10-trans-eicosatetraenoic acid (Samuelsson's HETE)“, in: J. Am. Chem. Soc., 1978, 100, S. 1942–1943; doi:10.1021/ja00474a058.
- ↑ a b P. Collins, R. Ferrier: Monosacharides – Their Chemistry and their Roles in Natural Products, Wiley, West Sussex 1995, ISBN 0-471-95343-1.
- ↑ Christopher R. Schmid, Jerry D. Bryant: D-(R)-Glycerinaldehyde Acetonide In: Organic Syntheses. 72, 1995, S. 6, doi:10.15227/orgsyn.072.0006; Coll. Vol. 9, 1998, S. 450 (PDF).
- ↑ András Lipták, János Imre, János Harangi, Pál Nánási, András Neszmélyi: „Chemo-, stereo- and regioselective hydrogenolysis of carbohydrate benzylidene acetals. Synthesis of benzyl ethers of benzyl α-D-, methyl β-D-mannopyranosides and benzyl α-D-rhamnopyranoside by ring cleavage of benzylidene derivatives with the LiAlH4-AlCl3 reagent“, in: Tetrahedron, 1982, 38, S. 3721–3727; doi:10.1016/0040-4020(82)80083-5.
- ↑ James A. Marshall, Joseph D. Trometer, Bruce E. Blough, Thomas D. Crute: „Stereochemistry of SN2' additions to acyclic vinyloxiranes“, in J. Org. Chem., 1988, 53, S. 4274–4282 doi:10.1021/jo00253a020.
- ↑ T. Tsunoda, M. Suzuki, R. Noyori: „A facile procedure for acetalization under aprotic conditions“, in: Tetrahedron Lett., 1980, 21, S. 1357–1358; doi:10.1016/S0040-4039(00)74575-8.
- ↑ Juji Yoshimura, Shigeomi Horito, Hiroriobu Hashimoto: „Facile Synthesis of 2,3,4,6-Tetra-O-benzyl-D-glucopyranosylidene Acetals Using Trimethylsilyl Trifluoromethanesulfonate Catalyst“, in: Chem. Lett., 1981, 10, S. 375–376; doi:10.1246/cl.1981.375.
- ↑ Bruce H. Lipshutz, Daniel Pollart, Joseph Monforte, Hiyoshizo Kotsuki: „Pd(II)-catalyzed acetal/ketal hydrolysis/exchange reactions“, in: Tetrahedron Lett., 1985, 26, S. 705–708; doi:10.1016/S0040-4039(00)89114-5.
- ↑ Kwan Soo Kim, Yang Heon Song, Bong Ho Lee, Chi Sun Hahn: „Efficient and selective cleavage of acetals and ketals using ferric chloride adsorbed on silica gel“, in: J. Org. Chem., 1986, 51, S. 404–407; doi:10.1021/jo00353a027.
- ↑ P.J. Kocieński: Protecting Groups, S. 167–170.
- ↑ P.J. Kocieński: Protecting Groups, S. 164–167.
- ↑ P.J. Kocieński: Protecting Groups, S. 176.
- ↑ P.J. Kocieński: Protecting Groups, S. 178–180.
- ↑ Samuel J. Danishefsky, Nathan B. Mantlo, Dennis S. Yamashita, Gayle. Schulte: „Concise route to the calichemicin-esperamicin series: the crystal structure of an aglycone prototype“, in: J. Am. Chem. Soc., 1988, 110, S. 6890–6891; doi:10.1021/ja00228a051.
- ↑ John N. Haseltine, Maria Paz Cabal, Nathan B. Mantlo, Nobuharu Iwasawa, Dennis S. Yamashita, Robert S. Coleman, Samuel J. Danishefsky, Gayle K. Schulte: „Total synthesis of calicheamicinone: new arrangements for actuation of the reductive cycloaromatization of aglycon congeners“, in: J. Am. Chem. Soc., 1991, 113, S. 3850–3866; doi:10.1021/ja00010a030.
- ↑ P.J. Kocieński: Protecting Groups, S. 119.
- ↑ Satomi Niwayama: „Highly Efficient Selective Monohydrolysis of Symmetric Diesters“, in: J. Org. Chem., 2000, 65, S. 5834–5836; doi:10.1021/jo0001986.
- ↑ J.M. Khurana, Arti Sehgal: „An efficent and convenient procedure for ester hydrolysis“, in: Org. Prep. Proced. Ind., 1994, 26, S. 580–583.
- ↑ Robert V. Stevens, Albert W. M. Lee: „Stereochemistry of the Robinson-Schoepf reaction. A stereospecific total synthesis of the ladybug defense alkaloids precoccinelline and coccinelline“, in: J. Am. Chem. Soc., 1979, 101, S. 7032–7035; doi:10.1021/ja00517a042.
- ↑ J. Wrobel, K. Takahashi, V. Honkan, G. Lannoye, J. M. Cook, Steven H. Bertz: „α-Lithio ketones. 1. Stereocontrolled synthesis of (±)-modhephene via the Weiss reaction“, in: J. Org. Chem., 1983, 48, S. 139–141; doi:10.1021/jo00149a034.
- ↑ Dennis D. Keith, John A. Tortora, Roxana Yang: „Synthesis of L-2-amino-4-methoxy-trans-but-3-enoic acid“, in: J. Org. Chem., 1978, 43, S. 3711–3713; doi:10.1021/jo00413a016.
- ↑ Peter Mohr, Nada Waespe-Šarčević, Christoph Tamm, Krystyna Gawronska, Jacek K. Gawronski: „A Study of Stereoselective Hydrolysis of Symmetrical Diesters with Pig Liver Esterase“, in: Helv. Chim. Acta, 1983, 66, S. 2501–2511; doi:10.1002/hlca.19830660815.
- ↑ Théophile Tschamber, Nada Waespe-Šarčević, Christoph Tamm: „Stereocontrolled Synthesis of an Epimer of the C(19)-to-C(27) Segment of Rifamycin S“, in: Helv. Chim. Acta, 1986, 69, S. 621–625; doi:10.1002/hlca.19860690311.
- ↑ Yves Rubin, Carolyn B. Knobler, Francois Diederich: „Precursors to the cyclo[n]carbons: from 3,4-dialkynyl-3-cyclobutene-1,2-diones and 3,4-dialkynyl-3-cyclobutene-1,2-diols to cyclobutenodehydroannulenes and higher oxides of carbon“, in: J. Am. Chem. Soc., 1990, 112, S. 1607–1617; doi:10.1021/ja00160a047.
- ↑ Sunggak Kim, Yong Gil Kim, Deog-il Kim: „A novel method for selective dioxolanation of ketones in the presence of aldehydes“, in: Tetrahedron Lett., 1992, 33, S. 2565–2566; doi:10.1016/S0040-4039(00)92243-3.
- ↑ G. Bauduin, D. Bondon, Y. Pietrasanta, B. Pucci: „Reactions de transcetalisation – II: Influence des facteurs steriques et electroniques sur les energies de cetalisation“, in: Tetrahedron, 1978, 34, S. 3269–3274; doi:10.1016/0040-4020(78)80243-9.
- ↑ John E. McMurry, Stephen J. Isser: „Total synthesis of longifolene“, in: J. Am. Chem. Soc., 1972, 94, S. 7132–7137; doi:10.1021/ja00775a044.
- ↑ M.P. Bosch, M. Pilar Bosch, Francisco Camps, Jose Coll, Angel Guerrero, Toshio Tatsuoka, Jerrold Meinwald: „A stereoselective total synthesis of (±)-muzigadial“, in: J. Org. Chem., 1986, 51, S. 773–784; doi:10.1021/jo00356a002.
- ↑ Ulrich Schmidt, Thomas Beuttler, Albrecht Lieberknecht, Helmut Griesser: „Aminosäuren und peptide – XXXXII. Synthese von Chlamydocin + epi-Chlamydocin“, in: Tetrahedron Lett., 1983, 24, S. 3573–3576; doi:10.1016/S0040-4039(00)88171-X.
- ↑ Elias J. Corey, Plato A. Magriotis: „Total synthesis and absolute configuration of 7,20-diisocyanoadociane“, in: J. Am. Chem. Soc., 1987, 109, S. 287–289; doi:10.1021/ja00235a052.
- ↑ Elias J. Corey, Kyriacos C. Nicolaou, Takeshi Toru: „Total synthesis of (±)-vermiculine“, in: J. Am. Chem. Soc., 1975, 97, S. 2287–2288; doi:10.1021/ja00841a058.
- ↑ Tainejiro Hiyama, Akihiro Kanakura, Hajime Yamamoto, Hitosi Nozaki: „General Route to α,β-unsaturated Aldehydes of Homoterpenoid and terpenoid Structure. Sythesis of JH-II and β-Sinensal“, in: Tetrahedron Lett., 1978, 19, S. 3051–3054; doi:10.1016/S0040-4039(01)94936-6.
- ↑ F. Huet, A. Lechevallier, M. Pellet, J.M. Conia: „Wet Silica Gel; A Convenient Reagent for Deacetalization“, in: Synthesis, 1978, S. 63–64.
- ↑ F. Zymalkokowski: Katalytische Hydrierung, Ferdinand Enke Verlag, Stuttgart 1965, S. 127–133.
- ↑ P.J. Kocieński: Protecting Groups, S. 136.
- ↑ P.J. Kocieński: Protecting Groups, S. 139–142.
- ↑ Ahmed M. Tafesh, Jens Weiguny: „A Review of the Selective Catalytic Reduction of Aromatic Nitro Compounds into Aromatic Amines, Isocyanates, Carbamates, and Ureas Using CO“, in: Chem. Rev., 1996, 96, S. 2035–2052; doi:10.1021/cr950083f.
- ↑ Evan L. Allred, Boyd R. Beck, Kent J. Voorhees: „Formation of carbon-carbon double bonds by the reaction of vicinal dihalides with sodium in ammonia“, in: J. Org. Chem., 1974, 39, S. 1426–1427; doi:10.1021/jo00926a024.
- ↑ Timothy S. Butcher, Feng Zhou, Michael R. Detty: „Debrominations of vic-Dibromides with Diorganotellurides. 1. Stereoselectivity, Relative Rates, and Mechanistic Implications“, in: J. Org. Chem., 1998, 63, S. 169–176; doi:10.1021/jo9713363.
- ↑ C. J. Li, David N. Harpp: „Bis(triphenylstanyl)telluride a mild and selective reagent for telluration and debromination“, in: Tetrahedron Lett., 1990, 31, S. 6291–6293; doi:10.1016/S0040-4039(00)97045-X.
- ↑ Corrado Malanga, Serena Mannucci, Luciano Lardicci: „Carbon-halogen bond activation by nickel catalyst: Synthesis of alkenes, from 1,2-dihalides“, in: Tetrahedron, 1998, 54, S. 1021–1028; doi:10.1016/S0040-4020(97)10203-4.
- ↑ Byung Woo Yoo, Seo Hee Kim, Jun Ho Kim: „A Mild, Efficient, and Selective Debromination of vic-Dibromides to Alkenes with Cp2TiCl2/Ga System“, in: Bull. Korean Chem. Soc., 2010, 31, S. 2757–2758; doi:10.5012/bkcs.2010.31.10.2757.
- ↑ Antonius J. H. Klunder, Jie Zhu, Binne Zwanenburg: „The Concept of Transient Chirality in the Stereoselective Synthesis of Functionalized Cycloalkenes Applying the Retro-Diels-Alder Methodology“, in: Chem. Rev., 1999, 99, S. 1163–1190; doi:10.1021/cr9803840.
- ↑ Hideyuki Tanaka, Takashi Kamikubo, Naoyuki Yoshida, Hideki Sakagami, Takahiko Taniguchi, Kunio Ogasawara: „Enantio- and Diastereocontrolled Synthesis of (−)-Iridolactone and (+)-Pedicularis-lactone“, in: Org. Lett., 2001, 3, S. 679–681; doi:10.1021/ol0070029.
- ↑ Martin Banwell, David Hockless, Bevyn Jarrott, Brian Kelly, Andrew Knill, Robert Longmore, Gregory Simpson: „Chemoenzymatic approaches to the decahydro-as-indacene cores associated with the spinosyn class of insecticide“, in: J. Chem. Soc., Perkin Trans. 1, 2000, S. 3555–3558; doi:10.1039/b006759h.
- ↑ Wenzel E. Davidsohn, Malcolm C. Henry: „Organometallic Acetylenes of the Main Groups III–V“, in: Chem. Rev., 1967, 67, S. 73–106; doi:10.1021/cr60245a003.
- ↑ Barry J. Teobald: „The Nicholas reaction: the use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis“, in: Tetrahedron, 2002, 58, S. 4133–4170; doi:10.1016/S0040-4020(02)00315-0.
- ↑ Kenneth M. Nicholas, R. Pettit: „An alkyne protection group“, in: Tetrahedron Lett., 1971, 37, S. 3475–3478; doi:10.1016/S0040-4039(01)97209-0.
- ↑ Richard E. Connor, Kenneth M. Nicholas: „Isolation, characterization, and stability of α-[(ethynyl)dicobalt hexacarbonyl] carbonium ions“, in: J. Organomet. Chem., 1977, 125, C45–C48; doi:10.1016/S0022-328X(00)89454-1.
- ↑ Rosa F. Lockwood, Kenneth M. Nicholas: „Transition metal-stabilized carbenium ions as synthetic intermediates. I. α-[(alkynyl)dicobalt hexacarbonyl] carbenium ions as propargylating agents“, in: Tetrahedron Lett., 1977, S. 4163–4166; doi:10.1016/S0040-4039(01)83455-9.
- ↑ K.M. Nicholas, R. Pettit: „On the stability of α-(alkynyl)dicobalt hexacarbonyl carbonium ions“, in: J. Organomet. Chem., 1972, 44, C21–C24; doi:10.1016/0022-328X(72)80037-8.
- ↑ T. Reichstein, A. Grüssner: „Eine ergiebige Synthese der L-Ascorbinsäure (C-Vitamin)“, in: Helv. Chim. Acta, 1934, 17, S. 311–328; doi:10.1002/hlca.19340170136.
- ↑ K.C. Nicolaou, E.J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, VCH Verlagsgesellschaft mbH, Weinheim 1996, S. 711–729, ISBN 3-527-29284-5.
- ↑ Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, 4th Ed., John Wiley & Sons Inc., Hoboken, New Jersey, S. 10–13; ISBN 0-471-69754-0.
- ↑ J.M. McClure, Samuel J. Danishefsky: „A novel Heck arylation reaction: rapid access to congeners of FR 900482“, in: J. Am. Chem. Soc., 1993, 115, S. 6094–6100; doi:10.1021/ja00067a026.
- ↑ Serge L. Beaucage, Radhakrishman P. Iyer: „Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach“, in: Tetrahedron, 1992, 48, S. 2223–2311; doi:10.1016/S0040-4020(01)88752-4.
- ↑ Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2194; doi:10.1002/ange.19961081805.
- ↑ Marco Eissen, Radoslaw Mazur, Heinz-Georg Quebbemann und Karl-Heinz Pennemann: „Atom Economy and Yield of Synthesis Sequences“, in: Helv. Chim. Acta, 2004, 87, S. 524–535; doi:10.1002/hlca.200490050.
- ↑ Marco Eissen, Jürgen O. Metzger: „Environmental Performance Metrics for Daily Use in Synthetic Chemistry“, in: Chemistry – A European Journal, 2002, 8, S. 3580–3585; doi:10.1002/chin.200247273.
- ↑ Marco Eissen, Jürgen O. Metzger, Eberhard Schmidt, Uwe Schneidewind: 10 Jahre nach „Rio“ – Konzepte zum Beitrag der Chemie zu einer nachhaltigen Entwicklung, in: Angewandte Chemie, 2002, 114, S. 402–425; doi:10.1002/1521-3757(20020201)114:3<402::AID-ANGE402>3.0.CO;2-D.
- ↑ Siegfried Hauptmann: Organische Chemie, 2. Auflage, VEB Deutscher Verlag für Grundstoffindustrie, Leipzig 1985, ISBN 3-342-00280-8, S. 621–622.
- ↑ Hideaki Muratake, Harumi Kumagami, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part 3 Total synthesis of (±)-hapalindoles H and U, in: Tetrahedron, 1990, 46, S. 6351–6360; doi:10.1016/S0040-4020(01)96007-7.
- ↑ Hideaki Muratake, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part I Total synthesis of (±)-hapalindoles J and M, in: Tetrahedron, 1990, 46, S. 6331–6342; doi:10.1016/S0040-4020(01)96005-3.
- ↑ Hideaki Muratake, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part 2. Lithium aluminum hydride reduction of the electron-rich carbon-carbon double bond conjugated with the indole nucleus, in: Tetrahedron, 1990, 46, S. 6343–6350; doi:10.1016/S0040-4020(01)96006-5.
- ↑ Phil S. Baran, Thomas J. Maimone, Jeremy M. Richter: Total synthesis of marine natural products without using protecting groups, in: Nature, 446, S. 404–408; doi:10.1038/nature05569.