Hemoglobina



Hemoglobina (gr. αἷμα haîma „krew”[1], łac. globus „kula”), oznaczana też skrótami Hb lub HGB – czerwony barwnik krwi, białko zawarte w erytrocytach, którego zasadniczą funkcją jest transportowanie tlenu – przyłączanie go w płucach i uwalnianie w tkankach. Prawie wszystkie kręgowce posiadają hemoglobinę[2], z wyjątkiem ryb z rodziny bielankowatych[3].
Wśród ssaków hemoglobina stanowi około 96% suchej masy i około 35% masy całkowitej (z uwzględnieniem wody) krwinki czerwonej[4]. Hemoglobina ma zdolność wiązania tlenu 1,34 ml O2 na gram, co zwiększa całkowitą pojemność tlenową krwi 70-krotnie w porównaniu do tlenu rozpuszczonego w samym osoczu krwi[5].
Mutacje genu hemoglobiny prowadzą do chorób dziedzicznych: anemii sierpowatej, talasemii lub rzadkich chorób zwanych hemoglobinopatiami[6].
Budowa hemoglobiny
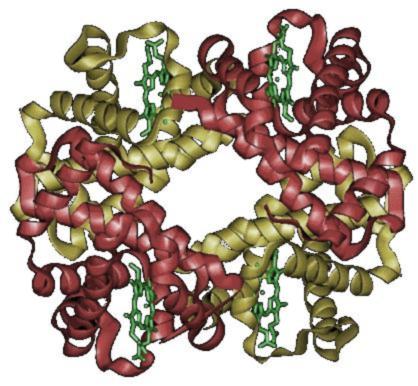
[edytuj | edytuj kod]Cząsteczka hemoglobiny jest tetramerem złożonym z dwóch par białkowych podjednostek. Podjednostki oznaczone są najczęściej literami greckiego alfabetu (np. α, β, γ, δ).
| Nazwa hemoglobiny | Pary podjednostek |
|---|---|
| HbA | α2β2 |
| HbF | α2γ2 |
| HbA2 | α2δ2 |
| HbS | α2S2 |
Podjednostki nie są związane kowalencyjnie. Każda podjednostka zawiera jako grupę prostetyczną (niebiałkową) cząsteczkę hemu. Cząsteczka hemu zawiera położony centralnie kation żelaza (Fe2+) umożliwiający jej wiązanie cząsteczek tlenu (O2). Jedna cząsteczka hemoglobiny może przyłączyć od jednej do czterech cząsteczek tlenu[7], co powoduje, że hemoglobina może występować albo w stanie „odtlenowanym” (deoxyHb) lub w różnym stopniu „utlenowania” (oxyHb). Hem nadaje białku (i krwi) czerwony kolor.
Przykładowa hemoglobina zbudowana z 574 reszt aminokwasowych ma masę cząsteczkową ok. 66,5 kDa, a jej wzór sumaryczny to C3032H4816O872N780S8Fe4[8].
Konformacja łańcuchów α i β ludzkiej hemoglobiny wykazuje podobieństwa do cząsteczki mioglobiny.
Podział hemoglobin
[edytuj | edytuj kod]Hemoglobiny „prawidłowe”
[edytuj | edytuj kod]- HbA (HbA1) (2α2β) – prawidłowa hemoglobina dorosłych
- HbA2 (2α2δ) – prawidłowa hemoglobina dorosłych; stanowi około 1,5–3% hemoglobiny
- HbF (2α2γ) – hemoglobina płodowa; ma większe powinowactwo do tlenu niż HbA, dzięki czemu jest w stanie pobrać tlen z krwi matki przez łożysko i uwolnić go w tkankach płodu. W życiu pozamacicznym jest zastępowana, gdyż słabiej uwalnia tlen w tkankach przy wyższym ciśnieniu parcjalnym tlenu. U dorosłych do 2%
- hemoglobiny embrionalne – mają podobne właściwości jak HbF:
- Hemoglobina Gower 1 (ξ2ε2)
- Hemoglobina Gower 2 (α2ε2)
- Hemoglobina Portland (ξ2γ2)
- HbA1c – HbA z przyłączoną nieenzymatycznie, trwale cząsteczką glukozy do N-końcowych aminokwasów łańcuchów globiny. Duże stężenie świadczy o proporcjonalnie podwyższonej glikemii, co może pozwolić na określenie średniego poziomu glukozy w surowicy przez okres 2-3 miesięcy, a to ma znaczenie np. w ocenie skuteczności leczenia cukrzycy. Norma stanowi 4–6% ogólnej ilości hemoglobiny. Produkty przejściowe pomiędzy HbA1 a HbA1c stanowią formy HbA1a oraz HbA1b będące zasadami Schiffa, w których glukoza jest przyłączona odwracalnie. Ogólna liczba wszystkich form glikowanej hemoglobiny powinna mieścić się w zakresie 6–8% ogólnej ilości hemoglobiny.
Hemoglobiny nieprawidłowe
[edytuj | edytuj kod]- HbS – jest efektem mutacji punktowej, w efekcie której następuje podmiana hydrofilowej reszty kwasu glutaminowego w pozycji A2 (6β) na hydrofobową resztę waliny, co powoduje powstanie lepkich miejsc i tworzenia agregatów nieutlenowanej HbS, które zniekształcają erytrocyty prowadząc do niedokrwistości sierpowatokrwinkowej.
- HbM – mutacja powodująca zamianę reszty histydyny w pozycji F8 na resztę tyrozyny, która stabilizuje żelazo w hemie w formie Fe3+ zamiast Fe2+. Hemoglobina zawierająca Fe3+ (methemoglobina, metHb) nie wiąże się z tlenem.
- Hemoglobina typu Chesapeake – zamiana Arg na Leu w pozycji FG4 (92 w łańcuchu α).
- Hemoglobina typu Bristol – zmiana Val na Asp w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji.
- Hemoglobina typu Sydney – zmiana Val na Ala w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji.
- Hemoglobina typu Hikari – zmiana Lys na Asn w pozycji 61 łańcucha β. Zmiana nie powoduje zaburzenia funkcji.
- Hemoglobina typu Milwaukee – zmiana Val na Glu w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji.
- Hemoglobina typu Lepore – (2α2Lepore) – hemoglobina w jednym z typów β-talasemii, wynik delecji genów kodujących łańcuchy β i δ. Ich resztki tworzą gen kodujący łańcuch Lepore.
Uwaga: litery greckie w nawiasach oznaczają jakie łańcuchy globiny wchodzą w skład cząsteczki.
Wiązanie tlenu
[edytuj | edytuj kod]Hemoglobina zawiera 4 grupy hemowe, dzięki czemu może związać i transportować 4 cząsteczki tlenu. O ile w wolnym hemie w obecności tlenu następuje szybkie utlenienie atomu żelaza(II) do żelaza(III), to w hemie związanym z globiną utlenienie takie nie następuje. Wynika to z otoczenia atomu żelaza przez niepolarne łańcuchy boczne reszt aminokwasowych globiny (podobny efekt występuje w mioglobinie). Ma to kluczowe znaczenie dla funkcjonowania hemoglobiny, gdyż jedynie forma zawierająca żelazo(II) jest zdolna do wiązania tlenu. Wiązanie to ma charakter koordynacyjny, a przyłączenie tlenu jest odwracalne. W hemoglobinie atom żelaza skoordynowany jest z czterema atomami azotu pierścienia porfirynowego oraz jednym białkowym atomem azotu reszty histydyny. Cząsteczka tlenu zajmuje szóstą pozycję koordynacyjną. Hemoglobina związana z tlenem nosi nazwę hemoglobiny utlenowanej lub oksyhemoglobiny, natomiast pozbawiona tlenu – deoksyhemoglobiny[9][10].
Przyłączenie cząsteczki tlenu do jednej z czterech cząstek hemu hemoglobiny powoduje zmianę struktury drugo-, trzecio- i czwartorzędowej całego tetrameru. Przyczyną jest wsunięcie atomu żelaza (położonego w przypadku nieutlenowanej hemoglobiny w odległości około 0,06 nm od płaszczyzny hemu) w płaszczyznę pierścienia hemu po połączeniu z tlenem.
Wsunięcie atomu żelaza pociąga związaną z nim tzw. histydynę proksymalną leżącą w pozycji F8, co powoduje przemieszczenie sąsiednich aminokwasów globiny. Doprowadza to w rezultacie do pęknięcia wiązań poprzecznych pomiędzy końcami karboksylowymi wszystkich czterech cząstek globiny. W efekcie dochodzi do rotacji pary α1/β1 względem pary α2/β2 o 15°.
Przyłączenie cząsteczki tlenu do hemoglobiny ułatwia przyłączanie następnych cząsteczek (tzw. wiązanie kooperacyjne), zaś odczepienie każdej cząsteczki tlenu ułatwia uwalnianie kolejnych cząsteczek O2. Wiązanie kooperacyjne sprzyja maksymalizacji wysycania tlenem hemoglobiny w płucach (przy danym ciśnieniu parcjalnym tlenu – PO2) oraz oddawania przez nią tlenu w tkankach.
Wiązanie dwutlenku węgla
[edytuj | edytuj kod]Hemoglobina transportuje około 15% z ogólnej ilości CO2 przenoszonego przez krew. W momencie gdy z cząsteczki hemoglobiny zostaje uwolniony tlen, dwutlenek węgla wchodzi w reakcję z grupą α-aminową globiny, tworząc karbaminian, jednocześnie w wyniku tej reakcji powstają wolne protony równoważące protony zużywane w płucach przez efekt Bohra (dwa mole protonów na każdy mol CO2). W wyniku powstania karbaminianu zmienia się ładunek grup na końcach łańcuchów globiny, co umożliwia tworzenie wiązań poprzecznych i przejście do stanu T.
W płucach natomiast, w momencie gdy dochodzi do przejścia ze stanu T do stanu R, w wyniku rozerwania wiązań poprzecznych uwolnione zostają protony z atomów azotu pierścieni imidazolowych histydyny znajdującej się w pozycji HC3 (His 146) na łańcuchach β. Łączą się one z wodorowęglanami, co prowadzi do powstania kwasu węglowego, rozkładanego następnie przez anhydrazę węglanową zawartą w erytrocytach na wodę i dwutlenek węgla usuwany z krążenia.
W tkankach występuje niższe pH niż w płucach, w związku z czym protony wiążą się z histydyną HC3, sprzyjając przejściu hemoglobiny ze stanu R do stanu T.
Rola bisfosfoglicerynianu
[edytuj | edytuj kod]W warunkach niedotlenienia w organizmie zwiększa się synteza 2,3-bisfosfoglicerynianu (BPG). Substancja ta, powstająca z 1,3-bisfosfoglicerynianu będącego produktem pośrednim glikolizy, ma właściwość wiązania się z tetramerem hemoglobiny znajdującym się w stanie T i stabilizowania go.
W stanie T konformacja łańcuchów hemoglobiny umożliwia wniknięcie między nie jednej cząsteczki BPG, która następnie wytwarza po trzy (w sumie sześć) wiązania poprzeczne z każdym z łańcuchów β, a dokładniej z posiadającymi dodatni ładunek resztami waliny w pozycji NA1, lizyny EF6 oraz histydyny H21. Te dodatkowe wiązania utrudniają przejście ze stanu T o niższym powinowactwie do tlenu do stanu R, w którym hemoglobina jest mniej skłonna do uwalniania tlenu.
Ciekawym przystosowaniem ewolucyjnym jest zamiana histydyny H21 na serynę w łańcuchu γ, który zastępuje łańcuch β w hemoglobinie płodowej HbF. Dzięki temu BPG ma mniejszy wpływ na hemoglobinę płodową, a co za tym idzie przy niższym stężeniu tlenu HbF ma do niego większe powinowactwo niż HbA. Efekt ten umożliwia wymianę gazową między krwią płodu a krwią matki zachodzącą w łożysku.
Normy
[edytuj | edytuj kod]
Normy ilości hemoglobiny we krwi dorosłego człowieka wynoszą około 11,0–17,5 g/dl, jednak ze względu na różne metody pomiarowe każde laboratorium analityczne ustala własne normy (zwykle podyktowane przez producenta analizatora). Ponadto fizjologicznie stężenie hemoglobiny u mężczyzn jest wyższe niż kobiet.
Stężenie hemoglobiny we krwi jest podstawowym kryterium przy diagnozowaniu niedokrwistości. Zmniejszone stężenie występuje również w stanach przewodnienia i w ciąży, a zwiększone świadczy o czerwienicy[11].
Pochodne hemoglobiny
[edytuj | edytuj kod]- oksyhemoglobina
- karbaminohemoglobina
- karboksyhemoglobina
- methemoglobina
- cyjanomethemoglobina
- sulfohemoglobina
Zobacz też
[edytuj | edytuj kod]Przypisy
[edytuj | edytuj kod]- ↑ hem, [w:] Encyklopedia PWN [online], Wydawnictwo Naukowe PWN [dostęp 2019-10-20].
- ↑ Anthea Maton, Human Biology and Health, Prentice Hall, 1993, ISBN 978-0-13-981176-0 [dostęp 2024-10-19] (ang.).
- ↑ Bruce D. Sidell, Kristin M. O'Brien, When bad things happen to good fish: the loss of hemoglobin and myoglobin expression in Antarctic icefishes, „The Journal of Experimental Biology”, 209 (Pt 10), 2006, s. 1791–1802, DOI: 10.1242/jeb.02091, ISSN 0022-0949, PMID: 16651546 [dostęp 2024-10-19].
- ↑ R.I. Weed, C.F. Reed, G. Berg, Is hemoglobin an essential structural component of human erythrocyte membranes?, „The Journal of Clinical Investigation”, 42 (4), 1963, s. 581–588, DOI: 10.1172/JCI104747, ISSN 0021-9738, PMID: 13999462 [dostęp 2024-10-19].
- ↑ Carl E. Rhodes, Deanna Denault, Matthew Varacallo, Physiology, Oxygen Transport, „StatPearls”, Treasure Island (FL): StatPearls Publishing, 2024, PMID: 30855920 [dostęp 2024-10-19].
- ↑ Hemoglobinopathies and Thalassemias [online], web.archive.org, 15 grudnia 2007 [dostęp 2024-10-23] [zarchiwizowane z adresu 2007-12-15].
- ↑ Linda S. Costanzo, Physiology, wyd. 4. ed, Board review series, Philadelphia: Lippincott Williams & Wilkins, 2007, ISBN 978-0-7817-7311-9 [dostęp 2024-10-19].
- ↑ Eldra Pearl Solomon, Linda R. Berg, Diana W. Martin, Claude A. Villee: Biologia. Wyd. 1. Warszawa: Multico Oficyna Wydawnicza, 1996, s. 68. ISBN 83-7073-090-6. OCLC 37964610.
- ↑ Białka: mioglobina i hemoglobína, [w:] Robert Kincaid Murray, Daryl K. Granner, Victor W. Rodwell, Biochemia Harpera ilustrowana, wyd. 6, Warszawa: Wydawnictwo Lekarskie PZWL, 2008, s. 53–58, ISBN 978-83-200-3573-5.
- ↑ Lubert Stryer, Biochemia, wyd. 2, Warszawa: PWN, 2003, s. 66–69, ISBN 83-01-13978-1.
- ↑ J. Kabata, B. Ochrem, A. Hellmann, Badania laboratoryjne i morfologiczne, [w:] Piotr Gajewski (red.), Interna Szczeklika, Kraków: Medycyna Praktyczna, Polski Instytut Evidence Based Medicine, 2020, ISBN 978-83-7430-627-0.