Organolithiumchemie
De organolithiumchemie is de tak van de organische chemie die zich bezighoudt met de studie van de organolithiumverbindingen, een organometaalverbinding met een directe covalente binding tussen een koolstof- en een lithium-atoom. Omdat lithium een tamelijk elektropositief element is, brengen de bindingselektronen een groot deel van de tijd rond het koolstofatoom door, dat daardoor een vrij grote partiële negatieve lading draagt. Effectief betekent dit dat het koolstofatoom zich als carbanion gedraagt en zowel een sterke base als een goed nucleofiel is.
Productie
[bewerken | brontekst bewerken]Op industriële schaal worden organolithiumverbindingen bereid door de reactie tussen een halogeenkoolstofverbindingen met metallisch lithium:
- R-X + 2 Li → R-Li + LiX[1]
Als zijreactie, meer bepaald bij de alkyljodiden, treedt de Wurtz-reactie op, waarbij een al gevormd molecuul R-Li reageert met een nog niet gereageerd molecuul R-X, waarbij een R-R koppelingsproduct gevormd wordt. Door alkylchloriden of bromiden te gebruiken wordt dit probleem grotendeels voorkomen.
Een tweede syntheseroute verloopt via de reactie van een alkylhalide of aryl-alkyl-sulfide, met een lithiumzout van een radicaal-anion, zoals lithiumnaftalide. De radicalaire anionen worden verkregen via de reactie van aromatische verbindingen (naftaleen) met metallisch lithium. Omdat de reactie van alkylhaliden met de radicalaire anionen veel sneller verloopt dan de directe reactie met lithium, maakt deze laatste syntheseroute de productie van een aantal bijzondere organolithiumverbindingen mogelijk.
Een derde route verloopt via de uitwisseling van metaal en halogeen tussen een organisch halide, meestal bromide of jodide, en een organolithiumverbinding. De laatste is dan vaak n-BuLi, s-BuLi of t-BuLi). Omdat het hier om een evenwichtsreactie gaat, is de reactie alleen succesvol als ofwel het gevormde lithiumreagens een stabieler carbanion heeft dan het uitgangsmateriaal, ofwel een van de reactieproducten eenvoudig uit het reactiemengsel verwijderd kan worden. Omdat dat laatste meestal door de lage temperatuur waarbij gewerkt wordt niet mogelijk is, beperkt deze manier van synthese zich tot vinyl-, aryl- en primaire alkyl-reagentia. Met name voor de bereiding van gefunctionaliseerde reagentia is deze route waardevol omdat de grovere reactieomstandigheden, al is het maar de hoge temperatuur, vermeden kunnen worden.
De vierde route naar organolithiumverbindingen verloopt ook via een uitwisseling, nu tussen twee organometaalverbindingen. Opnieuw is sprake van een evenwichtsreactie, waarbij het meest elektropositieve metaal eindigt bij de meest elektronegatieve organische groep. Een voorbeeld van deze reactie is de bereiding van vinyllithium uitgaande van tetravinyltin en fenyllithium. Vinyllithium is zeer moeilijk te bereiden via andere methoden.
Als laatste wordt gebruikgemaakt van het deprotoneren van een organische verbinding met een organolithiumverbinding. Hier is de facto sprake van een zuur-basereactie.
Structuur
[bewerken | brontekst bewerken]Organolithiumverbindingen hebben de neiging clusters te vormen, waarin lithium gecoördineerd is aan meer dan één koolstofatoom en ook de koolstofatomen coördineren met meerdere lithiumatomen. Er zijn drie algemene factoren aan te wijzen die invloed hebben op de vorming en de grootte van de clusters:
- De elektrostatische interactie tussen tegengesteld geladen atomen en atoomgroepen.
- De coördinatieruimte rond lithium waarin zowel moleculen van het oplosmiddel als Lewisbasen een plaats kunnen vinden.
- Sterische hindering in het koolwaterstofgedeelte kan de vorming van grotere clusters blokkeren, of bij afwezigheid van sterische hindering juist mogelijk maken.[2]
De lithiumatomen komen in deze clusters vaak in gelijkzijdige driehoeken voor.
In vaste toestand vormen 4 lithiumatomen bij methyllithium een tetraëder, waarbij op elk vlak een methylgroep zit die gelijkwaardig met alle drie de lithiumatomen gebonden is (η3-hapticiteit). Interacties over grotere afstanden tussen (MeLi)4 clusters zijn gebaseerd op η3-Li-CH3-η1-Li binding. Butyllithium vormt een hexameer: Li6-octaëder zonder lange-afstandinteracties.
Het toevoegen van een Lewisbase, zoals de oplosmiddelen di-ethylether, THF of stikstofliganden (TMEDA, PMDTA, sparteïne), heeft vaak tot gevolg dat de clusters afgebroken worden, waardoor ze beter oplosbaar worden en daarmee ook reactiever. Als vaste stof vormt methyllithium met (-)-sparteïne een dimeer. Het complex van butyllithium met PMDTA kan het best beschreven worden als monomeer butyllithium.
In oplossing vormt methyllithium tetrameren met THF. n-butyllithiumhexameren worden gevormd met benzeen of tetrameren met THF. t-BuLi vormt dimeren met THF. Isopropyllithium vormt in cyclopentaan een mengsel van hexameer, octameer en nonameer.
In de (zuur-base)reactie van n-butyllithium met fenylthioacetyleen bij −135 °C in THF kan via 7Li-NMR-spectroscopie een aantal verschillende aggregaten van de lithiumverbinding worden waargenomen:[3]
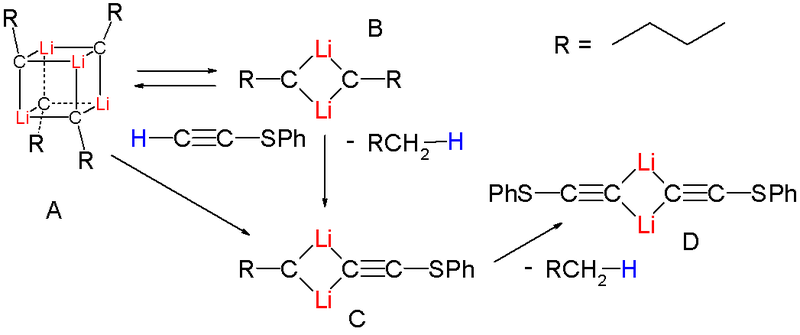
Het cubaan-achtige tetrameer A is nauwelijks reactief in vergelijking met het dimeer B, waaruit eerst gemengde dimeren C ontstaan en uiteindelijk homodimeer D. De versnellingsfactor voor het dimeer ten opzichte van het tetrameer is vastgestald op meer dan 3,2 × 108.
Toepassingen
[bewerken | brontekst bewerken]Organolithiumverbindingen zijn sterk gepolariseerd door het elektropositieve karakter van lithium (de elektronegativiteit van lithium bedraagt immers 1,0). Het gevolg hier is dat het reactieve nucleofielen zijn, die met vrijwel elk elektrofiel zullen reageren. Ze zijn vergelijkbaar met Grignardreagentia, maar dan veel reactiever. Ten gevolge van die reactiviteit is de aanwezigheid van (zelfs sporen) water, zuurstof (O2) en koolstofdioxide funest voor de bedoelde reactie. In aanwezigheid van water kan de R-groep geprotoneerd worden: deze reactie is thermodynamisch gezien zeer stabiel, waardoor de omgekeerde reactie nooit zal plaatsvinden. Organolithiumverbindingen worden daarom altijd in een beschermende atmosfeer (stikstof of argon) bewaard en ingezet. Tevens worden droge oplosmiddelen (droge ethers) gebruikt als reactiemidden.
De commercieel beschikbare organolithiumverbindingen, zoals n-BuLi, s-BuLi, t-BuLi, MeLi en PhLi, worden veel ingezet als zeer sterke basen. Organolithiumverbindingen kunnen bijna alle waterstofhoudende verbindingen deprotoneren, met uitzondering van de alkanen. In principe is de deprotonering compleet als de pKa van de te deprotoneren verbinding 2 eenheden groter is dan die van de gebruikte lithiumverbinding. In de praktijk geldt een groter verschil, omdat de kleine pKa ook meestal een lage reactiesnelheid betekent. Een groter verschil in pKa maakt de reactiesnelheid dan acceptabel. De basesterkte van de organolithiumverbindingen stijgt licht met een toenemend aantal alkylgroepen op het ladingdragende koolstofatoom. Dat maakt tert-butyllithium de sterkste, commercieel verkrijgbare base, met een pKa groter dan 53.
De metallatiereacties met organolithiumverbindingen vormen een belangrijk synthetisch hulpmiddel. Enige toepassingen staan hieronder:

Organolithiumverbindingen worden in de synthetisch organische chemie toegepast in addities over de carbonylgroep (het organische deel van de organolithiumverbinding koppelt aan koolstof, lithium aan zuurstof) of aan andere koolstof-elektrofielen. De basische zijreactie (deprotonering) kan een serieus probleem zijn bij enolisseerbare carbonylverbindingen, zeker met sterisch gehinderde organolithiumreagentia als t-butyllithium. Grignardreagentia zijn minder reactief in de additie-reactie, maar geven ook minder problemen met deprotoneren.
Een belangrijke toepassing van organolithiumreagentia is de synthese van andere organometaalverbindingen, vaak met behulp van metaalhalides. Synthetisch belangrijk is de vorming van organokoper-verbindingen (zoals het Gilmanreagens) via de reactie van RLi met CuI of CuBr, en de bereiding van organozink-verbindingen door reactie met ZnCl2. Soms worden zelfs Grignardreagentia via RLi met MgBr2 gemaakt, vooral in gevallen waarin het lithiumreagens simpel te maken is, en de Grignard problemen geeft. organotin-, organosilicon-, organoboor-, organofosfor- en organozwavel-verbindingen ontstaan uit RLi met de juiste elektrofielen.
Een kortgeleden gepubliceerd overzicht (review) met betrekking tot de procesindustrie geeft aan dat de volgende organolithiumverbindingen het meest gebryuikt worden: n-Butyllithium, hexyllithium, s-butyllithium en fenyllithium.[4] Ook Methyllithium wordt veel toegepast. Twee van de meest gebruikte sterke bases die met behulp van nBuKi worden bereid zijn lithiumdi-isopropylamide (LDA) en lithiumhexamethyldisilazide (LiHMDS).
Aryllithium-derivaten zijn belangrijke tussenstoffen in orthometallering zoals die optreedt bij de synthese van Me2NCH2C6H4-2-Li uit dimethylbenzylamine en nBuLi.
Reactiviteit
[bewerken | brontekst bewerken]Enige algemene reacties van organolithiumverbindingen zijn:
- Zij worden geprotoneerd in reacties met elke stof die zure protonen bevat.
- De reactie met ketonen en aldehyden waarbij alcoholen ontstaan.[5]
- Reacties met carbonzuren of zuurchlorides waarbij ketonen worden gevormd.[6]
- Reacties met esters waarbij tertiaire alcoholen ontstaan. In het genoemde voorbeeld bleek ethyllithium erg effectief, terwijl ethylmagnesiumbromide vooral reductie te gaf.[7]

- Reacties met oximen tot de overeenkomstige amines.[5]
- Reacties met isocyanides naar lithiumaldimines. De op de koppeling volgende hydrolyse zet het organolithiumreagens om in zijn homologe aldehyde.[8]
- Reactie met een aantal epoxides tot alkenen.[9]
Stabiliteit
[bewerken | brontekst bewerken]Organolithiumreagentia reageren ook met de ethers die als oplosmiddel gebruikt worden. In de tabel hieronder zijn een aantal halveringstijden verzameld van een aantal veel gebruikte organolithiumverbindingen in combinatie met veel gebruikte oplosmiddelen.[10] De genoemde tijden vormen een indicatie, geen absolute meetwaarden.
| Oplosmiddel / Temperatuur | n-BuLi | s-BuLi | t-BuLi | MeLi | CH2=C(OEt)-Li | CH2=C(SiMe3)-Li |
|---|---|---|---|---|---|---|
| THF / −20 °C | 40 min, 360 min | |||||
| THF / 20 °C | >15 u | 17 u | ||||
| THF / 35 °C | 10 min | |||||
| THF / TMEDA / −20 °C | 55 u | |||||
| THF / TMEDA / 0 °C | 340 min | |||||
| THF / TMEDA / 20 °C | 40 min | |||||
| Ether / −20 °C | 480 min | |||||
| Ether / 0 °C | 61 min | |||||
| Ether / 20 °C | 153 u | < 30 min | 17 dagen | |||
| Ether / 35 °C | 31 u | |||||
| Ether / TMEDA / 20 °C | 603 min | |||||
| DME / −70 °C | 120 min | 11 min | ||||
| DME / −20 °C | 110 min | 2 min | << 2 min | |||
| DME / 0 °C | 6 min |
Zie ook
[bewerken | brontekst bewerken]- ↑ Stent, M. (2002). Generation of a Highly Basic and Nucleophilic Organolithium; Isopropyllithium. SyntheticPages (195).
- ↑ Structure Formation Principles and Reactivity of Organolithium Compounds Viktoria H. Gessner, Christian Daschlein, and Carsten Strohmann Chem. Eur. J. 2009, 15, 3320 – 3334 DOI:10.1002/chem.200900041
- ↑ Amanda C. Jones, Aaron W. Sanders, Martin J. Bevan, and Hans J. Reich (2007). Reactivity of Individual Organolithium Aggregates: A RINMR Study of n-Butyllithium and 2-Methoxy-6-(methoxymethyl)phenyllithium (Communication). J. Am. Chem. Soc. 129 (12): 3492–3493. PMID 17341084. DOI: 10.1021/ja0689334.
- ↑ Wu, G. Huang, M. (2006). Organolithium Reagents in Pharmaceutical Asymmetric Processes. Chemical Reviews 106 (7): 2596–2616. PMID 16836294. DOI: 10.1021/cr040694k.
- ↑ a b Advanced Organic Chemistry F.A. Carey R.J. Sundberg 2nd Edition ISBN 0-306-41088-5
- ↑ (en) T.M. Bare & H.O. House (1973) - Methyl Ketones from Carboxylic Acids: Cyclohexyl Methyl Ketone, Organic Syntheses, 5, p. 775
- ↑ Posner, Gary H., Jae Kyoo Lee, Qiang Wang, Sara Peleg, Martin Burke, Henry Brem, Patrick Dolan, Thomas W. Kensler (1998). Noncalcemic, Antiproliferative, Transcriptionally Active, 24-Fluorinated Hybrid Analogues of the Hormone 1α,25-Dihydroxyvitamin D3. Synthesis and Preliminary Biological Evaluation. Journal of Medicinal Chemistry 41 (16): 3008–3014. PMID 9685240. DOI: 10.1021/jm980031t.
- ↑ (en) G.E. Niznik, W.H. Morrison & H.M. Walborsky (1988) - 1-d-Aldehydes from Organometallic Reagents: 2-Methylbutanal-1-d, Organic Syntheses, 6, p. 751
- ↑ (en) D.M. Hodgson, P.G. Humphreys & M.J. Fleming (2008) - Organolithiums and Lithium 2,2,6,6-Tetramethylpiperidide in Reductive Alkylation of Epoxides: Synthesis of (E)-Alkenes, Organic Syntheses, 85, p. 1–9
- ↑ Stanetty, P.; Koller, H.; Mihovilovic, M. (1992). Directed Ortho-Lithiation of Phenylcarbamic Acid 1,l-Dimethylethyl Ester (N-Boc-aniline). Revision and Improvements. J. Org. Chem. 57: 6833–6837. DOI: 10.1021/jo00051a030.