Distrofia corneale
| Distrofia corneale | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RFG140 |
| Specialità | neurologia |
| Classificazione e risorse esterne (EN) | |
| ICD-10 | H18.5 |
| MeSH | D003317 |
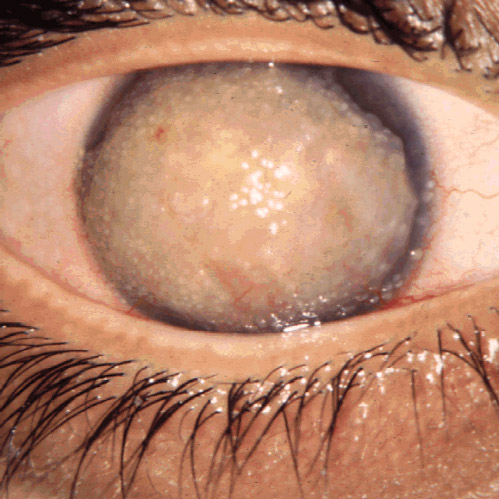
Con distrofia corneale si indica un gruppo eterogeneo di disturbi di natura non infiammatoria, progressivi, non correlati a fattori ambientali, che colpiscono la cornea. Si tratta di disturbi solitamente determinati geneticamente, bilaterali e simmetrici, non associati a disturbi sistemici che causano opacizzazione con conseguente riduzione dell'acuità visiva[1][2][3]. Il termine distrofia corneale è impreciso, ma continua ad essere utilizzato per il suo valore clinico[2] anche se per molti potrebbe avere un significato più storico che pratico[1][4].
Le distrofie corneali sono state individuate nell'uomo, ma anche nel cane e, più raramente, nel gatto.
Definizione e caratteristiche
[modifica | modifica wikitesto]Il termine distrofia corneale fu introdotto nel 1890 da Groenouw che riportò il caso di 2 pazienti con quelli che definì noduli cornea riferendosi probabilmente ad una distrofia granulare ed a una distrofia maculare. Successivamente Biber chiamò distrofia corneale un caso di distrofia tipo lattice. In seguito il termine fu utilizzato da Fuchs, Uhthoff e Yoshida.[1] La definizione è oggetto di controversia e da alcuni il termine “distrofia” è considerato in certi casi fuorviante visto che alcune forme si manifestano più come malattie degenerative che ereditarie. Inoltre anche in alcune di quelle di cui è stato individuata l'associazione ad un preciso locus non è stato identificato il gene o non si è compreso il carattere di ereditarietà (in genere autosomica dominante o recessiva)[1]. Alcune sono asintomatiche o unilaterali oppure si presentano associate a disturbi sistemici. Sono tipicamente caratterizzate da un’alterazione morfofunzionale conseguente a modificazioni del normale trofismo corneale[3].
Nomenclatura e classificazione
[modifica | modifica wikitesto]
La natura eterogenea dei disturbi oltre che l'utilizzo fuorviante del termine “distrofia corneale” attribuito a disturbi degenerativi e la tendenza a individuare e denominare nuove forme di distrofia corneale prima che fosse accertata e condivisa l'effettiva esistenza di una nuova malattia, ha generato molte incomprensioni ed in certi casi errate diagnosi[1][5].
Per dirimere le controversie sulla classificazione e nomenclatura delle distrofie corneali si è costituito l’International Committee for Classification of Corneal Dystrophies (IC3D) che nel 2008 ha proposto una classificazione, poi aggiornata nel 2015. Le distrofie della cornea si possono classificare clinicamente in gruppi, in base alla localizzazione anatomica dell'anomalia. Alcune colpiscono soprattutto l'epitelio corneale, la sua membrana basale o lo strato di Bowman e lo stroma corneale superficiale (distrofie corneali anteriori), lo stroma corneale (distrofie corneali stromali), o la membrana di Descemet e l'endotelio corneale (distrofie corneali posteriori)[4]. Nella revisione del IC3D è stata superata una classificazione troppo dipendente da specifici strati corneali, raggruppando le distofie corneali in epiteliali e subepiteliali, stromali, endoteliali. Le diverse distrofie che possono colpire diversi strati ma dipendono da un unico gene (TGFBI) e locus vengono raggruppate in un quarto gruppo definito epiteliale-stromale[1]. I recenti progressi della genetica molecolare hanno identificato molti dei difetti genetici responsabili della maggior parte delle distrofie corneali[4][6], ma la rapida e continua evoluzione delle informazioni sulle basi genetiche delle distrofie corneali oltre che alcune distrofie dipendenti da decine di mutazioni[7], hanno portato nel 2015 alla decisione dell'I3CD di non specificare gene e locus delle diverse distrofie.
| Classificazione IC3D (2015) delle Distrofie Corneali[1] | Basi genetiche[1][4] | ||||
|---|---|---|---|---|---|
| Gruppo | Distrofia corneale | Categoria | Ereditarietà | Locus | Gene |
| Epiteliali e subepiteliali | |||||
| della membrana basale epiteliale (EBMD) | 1 in casi rari | per lo più degenerativa, sporadica | 5q31 | TGFBI in casi rari | |
| delle erosioni epiteliali ricorrenti (ERED) e varianti: Franceschetti(FRCD), Smolandiensis(DS), Helsinglandica(DH). | 4-3 | Autosomica dominante | Sconosciuto | Sconosciuto | |
| Mucinosa subepiteliale (SMCD) | 4 | Autosomica dominante | Sconosciuto | Sconosciuto | |
| di Meesmann (MECD) | 1 | Autosomica dominante | 12q12, 17q12 | KRT3, KRT12 | |
| Epiteliale di Lisch (LECD) | 2 | cromosoma X dominante | Xp 22.3 | Sconosciuto | |
| Gelatinosa a goccia (GDCD) | 1 | Autosomica recessiva | Ip32 | TACSTD2 | |
| TGFBI epiteliali-stromali | |||||
| di Reis-Bucklers (RBCD) | 1 | Autosomica dominante | 5q31 | TGFBI | |
| di Thiel-Benke (TBCD) | 2 | Autosomica dominante | 10q24 | Sconosciuto | |
| Reticolare (Lattice), tipo I (LCDI) | 1 | Autosomica dominante | 5q31 | TGFBI | |
| variante (III, IIIA, I/IIIA, IV) della LCDI | 1 | Autosomica dominante | 5q31 | TGFBI | |
| Granulare tipo I (GCDI) | 1 | Autosomica dominante | 5q31 | TGFBI | |
| Granulare tipo II (GCD2) – di Avellino | 1 | Autosomica dominante | 5q31. | TGFBI | |
| Stromali | |||||
| Maculare (MCD) | 1 | Autosomica recessiva | 16Q22 | CHST6 | |
| di Schnyder (SCCD) | 1 | Autosomica dominante | 1p36 | UBIAD1 | |
| Congenita stromale (CSCD) | 1 | Autosomica dominante | 12q31.33 | DCN | |
| di Fleck (FCD) | 1 | Autosomica dominante | 2q35 | PIP5K3 | |
| Posteriore amorfa (PACD) | 3 | Autosomica dominante | Sconosciuto | Sconosciuto | |
| della nebulosa centrale di Francois (CCDF) | 4 | Sconosciuta | Sconosciuto | Sconosciuto | |
| Pre-Descemetica | 1-4 | Sconosciuta | Sconosciuto | Sconosciuto | |
| Endoteliali | |||||
| di Fuchs (FECD) a comparsa tardiva | 1-2-3 | Sconosciuta, a volte Autosomica dominante | 13pter-q12.13 (FECD2), 18q21.2-q21.3 (FECD3), 20p13-p12 (FECD4), 5q33.1-q35.2 (FECD5), 10p11.2 (FECD6), 9p24.1-p22.1 (FECD7), 15q25 (FECD8). |
Sconosciuto,TCF8, SLC4A11 | |
| di Fuchs (FECD) a comparsa precoce | 1 | Autosomica dominante | Ip34.3 | COL8A2 | |
| Posteriore polimorfa (PPCD) 1 | 2 | Autosomica dominante | 20p11.12-q11.2 | Sconosciuto | |
| Posteriore polimorfa (PPCD) 2 | 1 | Autosomica dominante | Ip34.3-p32.3 | COL8A2 | |
| Posteriore polimorfa (PPCD) 3 | 1 | Autosomica dominante | 10p11.12 | ZEB1 | |
| Endoteliale congenita ereditaria (CHED) | 2 | Autosomica dominante | 20p11.12-q11.2 | Sconosciuto | |
| Endoteliale legata al cromosoma X (XECD) | 2 | cromosoma X dominante | Xq25 | Sconosciuto | |
| Legenda: – Categoria 1: distrofia ben definita, di cui un gene specifico responsabile è stato mappato e identificato– Categoria 2: distrofia ben definita, di cui un gene è stato mappato in uno o più loci cromosomici specifici, ma altri geni non sono ancora stati individuati– Categoria 3: distrofia ben definita, i cui geni responsabili non sono ancora stati mappati– Categoria 4: distrofia nuova, o documentata in passato, in cui l’evidenza che si tratti di un’entità distinta non è del tutto convincente[1]– TGFBI: gene del fattore di crescita trasformante beta indotto. | |||||
Le distrofie corneali di categoria 4, emergendo nuove prove in grado di distinguerle dalle altre distrofie, possono passare ad una categoria inferiore. In caso contrario possono venir cancellate, come è stato deciso dal IC3D per la distrofia di Grayson-Wilbrandt[1][8] .
Diagnosi
[modifica | modifica wikitesto]Il sospetto di distrofia corneale sorge nel caso di perdita della trasparenza corneale o di opacizzazione spontanea della cornea, soprattutto se bilaterale, e qualora siano presenti altri soggetti affetti nella famiglia. La diagnosi clinica si basa sull'età di esordio e sull'aspetto clinico della cornea al biomicroscopio con lampada a fessura. Per definire il tipo specifico di distrofia o degenerazione è necessario l'esame con microscopia ottica e microscopia elettronica a trasmissione (TEM) del tessuto corneale. Nei sottotipi di distrofie di cui è nota la mutazione genetica si può eseguire l'analisi molecolare per confermare la diagnosi[2].
Diagnosi differenziale
[modifica | modifica wikitesto]In assenza di test genetici e elementi di contesto (famigliarità) la diagnosi differenziale può risultare complessa dovendosi confrontare con le gammopatie monoclonali, l'amiloidosi, il deficit di lecitina-colesterolo-aciltransferasi, la malattia di Fabry, la cistinosi, la tirosinemia tipo 2, le malattie sistemiche da accumulo lisosomiale (mucoplisaccaridosi, lipidosi, mucolipidosi) e varie malattie della cute (ittiosi X-linked, cheratosi follicolare spinulosa decalvante)[2].
Note
[modifica | modifica wikitesto]- ^ a b c d e f g h i j Weiss Jayne S.; Møller Hans Ulrik, Aldave, Anthony J., Seitz Berthold, Bredrup Cecilie, Kivelä Tero, Munier Francis L., Rapuano Christopher J., Nischal Kanwal K., Kim Eung Kweon, Sutphin John, Busin Massimo, Labbé Antoine, Kenyon Kenneth R., Kinoshita Shigeru, Lisch Walter, IC3D Classification of Corneal Dystrophies—Edition 2, vol. 34, n. 2, febbraio 2015.
- ^ a b c d ORPHANET: Distrofia corneale
- ^ a b Unità Clinica Operativa CLINICA OCULISTICA – Friuli Venezia Giulia - DISTROFIE CORNEALI EREDITARIE (PDF), su aots.sanita.fvg.it. URL consultato il 5 febbraio 2017 (archiviato dall'url originale il 5 febbraio 2017).
- ^ a b c d Gordon K Klintworth, Corneal dystrophies, in Orphanet Journal of Rare Diseases, vol. 4, n. 7, febbraio 2009.
- ^ Weiss JS., Schnyder crystalline dystrophy sine crystals. Recommendation for a revision of nomenclature., in Ophthalmology, vol. 103, 1996, pp. 465–473.
- ^ N.Pescosolido, M.Autolitano – Le distrofie corneali
- ^ IC3D (2008) :Table of Genes and Mutations Associated with the Corneal Dystrophies
- ^ ORPHANET:distrofia corneale di Grayson-Wilbrandt
Voci correlate
[modifica | modifica wikitesto]- Cornea
- Occhio
- Ereditarietà autosomica dominante
- Ereditarietà autosomica recessiva
- Dominanza (genetica)
Altri progetti
[modifica | modifica wikitesto]Wikimedia Commons contiene immagini o altri file su distrofia corneale

| Controllo di autorità | Thesaurus BNCF 53659 |
|---|