Ecuación cinética
En química, la ley de velocidad o ecuación cinética es una expresión matemática empírica para la velocidad de reacción de una reacción particular en la cual solo aparecen concentraciones de especies químicas y parámetros constantes (normalmente coeficientes de velocidad y órdenes parciales de reacción).[1] Para muchas reacciones, la velocidad inicial viene dada por una ley potencial como la siguiente
donde y expresan la concentración de las especies
y , normalmente en moles por litro (molaridad, ). Los exponentes e son los órdenes parciales de reacción para
y siendo el orden de reacción global la suma de los exponentes. Suelen ser enteros positivos, pero también pueden ser cero, fraccionarios o negativos. El orden de reacción es un número que cuantifica el grado en que la velocidad de una reacción química depende de las concentraciones de los reactivos.[2] En otras palabras, el orden de reacción es el exponente al que se eleva la concentración de un reactivo concreto.[2] La constante es la constante de velocidad de reacción o coeficiente de velocidad denominándose a veces constante de velocidad o velocidad específica de reacción. Su valor puede depender de condiciones como la temperatura, la fuerza iónica, la superficie de un adsorbente o la irradiación de luz. Si la reacción llega a completarse, la ecuación de velocidad de la velocidad de reacción se aplica durante toda la reacción.
Las reacciones elementales (de un solo paso) y los pasos de reacción tienen órdenes de reacción iguales a los coeficientes estequiométricos de cada reactivo. El orden de reacción global, es decir, la suma de los coeficientes estequiométricos de los reactivos, es siempre igual a la molecularidad de la reacción elemental. Sin embargo, las reacciones complejas (de varios pasos) pueden tener o no órdenes de reacción iguales a sus coeficientes estequiométricos. Esto implica que el orden y la ecuación cinética de una reacción dada no pueden deducirse de forma fiable a partir de la estequiometría y deben determinarse experimentalmente, ya que un mecanismo de reacción desconocido podría ser elemental o complejo. Cuando se ha determinado la ecuación cinética experimental, suele ser útil para deducir el mecanismo de reacción.
La ecuación cinética de una reacción con un supuesto mecanismo de múltiples pasos a menudo puede derivarse teóricamente utilizando suposiciones de estado cuasi-estacionario a partir de las reacciones elementales subyacentes, y compararse con la ecuación cinética experimental como prueba del supuesto mecanismo. La ecuación puede implicar un orden fraccionario y depender de la concentración de una especie intermedia.
Una reacción también puede tener un orden de reacción indefinido con respecto a un reactivo si la velocidad no es simplemente proporcional a alguna potencia de la concentración de ese reactivo; por ejemplo, no se puede hablar de orden de reacción en la ecuación cinética de una reacción bimolecular entre moléculas adsorbidas:
Definición
[editar]Consideremos una reacción química típica en la que dos reactivos A y B se combinan para formar un producto C:
También se puede escribir
Los prefactores -1, -2 y 3 (con signo negativo para los reactivos porque se consumen) se conocen como coeficientes estequiométricos. Una molécula de A se combina con dos de B para formar 3 de C, por lo que si utilizamos el símbolo [X] para el número de moles de la sustancia química X,[3]
Si la reacción tiene lugar en un sistema cerrado a temperatura y volumen constantes, sin acumulación de productos de reacción intermedios, la velocidad de reacción se define como
donde es el coeficiente estequiométrico para el compuesto , con signo negativo para un reactivo.
La velocidad de reacción inicial
tiene cierta dependencia funcional de las concentraciones de los reactivos,
y esta dependencia se conoce como ecuación cinética o ley de velocidad.[4] Por lo general, esta ley no puede deducirse de la ecuación química y debe determinarse experimentalmente.[5]
Leyes potenciales
[editar]Una forma común para la ecuación cinética es una ley potencial:[5]
La constante se denomina constante de velocidad. Los exponentes, que pueden ser fraccionarios,[5] se llaman órdenes parciales de reacción y su suma es el orden global de reacción.[6]
Para una disolución diluida se ha comprobado empíricamente que una reacción elemental (con un único paso y un único estado de transición) obedece a la ley de acción de masas. Ésta predice que la velocidad depende únicamente de las concentraciones de los reactivos, elevadas a las potencias de sus coeficientes estequiométricos.[7]
Determinación del orden de reacción
[editar]Método de las velocidades iniciales
[editar]El logaritmo natural de la ecuación de la ley potencial es
Esto puede utilizarse para estimar el orden de reacción de cada reactivo. Por ejemplo, la velocidad inicial puede medirse en una serie de experimentos con diferentes concentraciones iniciales del reactivo mantienendo constantes las demás , de modo que
La pendiente de la gráfica de en función de corresponde entonces al orden con respecto al reactivo .[8][9]
Sin embargo, este método no siempre es fiable porque
- La medición de la velocidad inicial requiere la determinación precisa de pequeños cambios en la concentración en tiempos cortos (en comparación con la vida media de la reacción) y es sensible a errores, y
- la ecuación de la velocidad no se determinará completamente si la velocidad también depende de sustancias no presentes al principio de la reacción, como productos intermedios u otros productos.
Método integral
[editar]Por lo tanto, la ecuación cinética provisional determinada por el método de las velocidades iniciales se verifica normalmente comparando las concentraciones medidas durante un tiempo más largo (varias semividas) con la forma integrada de la ecuación de velocidad; esto supone que la reacción se completa.
Por ejemplo, la ley de velocidad integrada para una reacción de primer orden es
donde es la concentración en el instante y es la concentración inicial. La ley de velocidad de primer orden se confirma si es de hecho una función lineal del tiempo. En este caso la constante de velocidad es igual a la pendiente con el signo invertido.[10][11]
Método por exceso
[editar]El orden parcial con respecto a un reactivo dado puede evaluarse mediante el método por exceso (o de aislamiento) de Ostwald. En este, la concentración de un reactivo se mide con todos los demás presentes en gran exceso para que su concentración permanezca esencialmente constante. Para una reacción con ley de velocidad el orden parcial con respecto a se determina utilizando un gran exceso de . En este caso con y puede determinarse por el método integral. El orden con respecto a en las mismas condiciones (con en exceso) se determina mediante una serie de experimentos similares con un rango de concentración inicial de modo que se pueda medir la variación de .[12]
Orden cero
[editar]En las reacciones de orden cero la velocidad de reacción es independiente de la concentración de un reactivo, de modo que el cambio de su concentración no tiene ningún efecto sobre la velocidad de reacción. Así, la concentración cambia linealmente con el tiempo. Esto puede ocurrir cuando existe un cuello de botella que limita el número de moléculas de reactivo que pueden reaccionar al mismo tiempo, por ejemplo, si la reacción requiere el contacto con una enzima o una superficie catalítica.[13]
Muchas reacciones catalizadas por enzimas son de orden cero, siempre que la concentración del reactivo sea mucho mayor que la concentración de la enzima que controla la velocidad, de modo que la enzima esté saturada. Por ejemplo, la oxidación biológica del etanol a acetaldehído por la enzima alcohol deshidrogenasa hepática (LADH) es de orden cero en etanol.[14]
Del mismo modo, las reacciones con catálisis heterogénea pueden ser de orden cero si la superficie catalítica está saturada. Por ejemplo, la descomposición del fosfano (PH3) en una superficie de tungsteno caliente a alta presión es de orden cero en el fosfano, que se descompone a una velocidad constante.[13]
En la catálisis homogénea, el comportamiento de orden cero puede deberse a la inhibición reversible. Por ejemplo, la polimerización por metátesis de apertura en anillo utilizando un catalizador Grubbs de tercera generación muestra un comportamiento de orden cero en el catalizador debido a la inhibición reversible que se produce entre la piridina y el centro de rutenio.[15]
Primer orden
[editar]Una reacción de primer orden depende de la concentración de un solo reactivo (reacción unimolecular). Puede haber otros reactivos, pero su concentración no influye en la velocidad. La ley de velocidad de una reacción de primer orden es
Aunque no afecta a las matemáticas anteriores, la mayoría de las reacciones de primer orden se producen mediante colisiones intermoleculares. Estas colisiones, que aportan la energía al reactivo, son necesariamente de segundo orden. Sin embargo, la velocidad de estas colisiones queda enmascarada por el hecho de que el paso determinante de la velocidad sigue siendo la descomposición unimolecular del reactivo energizado.
La semivida es independiente de la concentración inicial y viene dada por . La vida media es τ = 1/k.[16]
Ejemplos de este tipo de reacciones son:
En química orgánica, la clase de reacciones SN1 (unimoleculares de sustitución nucleofílica) consiste en reacciones de primer orden. Por ejemplo, en la reacción de iones arildiazonio con nucleófilos en solución acuosa, , la ecuación de velocidad es donde Ar indica un grupo arilo.[20]
Segundo orden
[editar]Se dice que una reacción es de segundo orden cuando el orden global es dos. La velocidad de una reacción de segundo orden puede ser proporcional a una concentración al cuadrado,
o (más comúnmente) al producto de dos concentraciones, Como ejemplo del primer tipo, la reacción es de segundo orden en el reactivo y de orden cero en el reactivo . La velocidad observada viene dada por y es independiente de la concentración de CO.[21]
Para la velocidad proporcional a una concentración única al cuadrado, la dependencia temporal de la concentración viene dada por
La dependencia temporal para una velocidad proporcional a dos concentraciones desiguales es
si las concentraciones son iguales, satisfacen la ecuación anterior.
El segundo tipo incluye reacciones nucleófilas de adición-eliminación, como la hidrólisis alcalina del acetato de etilo:
Esta reacción es de primer orden en cada reactivo y de segundo orden en su conjunto:
Si la misma reacción de hidrólisis es catalizada por imidazol, la ecuación de velocidad se convierte en[20]
La velocidad es de primer orden en un reactivo (acetato de etilo), y también de primer orden en imidazol, que como catalizador no aparece en la ecuación química global.
Otra clase bien conocida de reacciones de segundo orden son las reacciones SN2 (sustitución nucleofílica bimolecular), como la reacción del bromuro de n-butilo con yoduro sódico en acetona:
Este mismo compuesto puede someterse a una reacción de eliminación bimolecular (E2), otro tipo común de reacción de segundo orden, si el yoduro sódico y la acetona se sustituyen por terbutóxido sódico como sal y terbutanol como disolvente:
Pseudo primer orden
[editar]Si la concentración de un reactivo permanece constante (porque es un catalizador, o porque está en gran exceso con respecto a los otros reactivos), su concentración puede incluirse en la constante de velocidad, lo que conduce a una ecuación de velocidad de pseudo-primer orden (u ocasionalmente de pseudo-segundo orden). Para una reacción típica de segundo orden con ecuación de velocidad si la concentración del reactivo B es constante, entonces donde la constante de velocidad de pseudo-primer orden La ecuación de velocidad de segundo orden se ha reducido a una ecuación de velocidad de pseudoprimer orden, lo que facilita mucho el tratamiento para obtener una ecuación de velocidad integrada.
Una forma de obtener una reacción de pseudo-primer orden es utilizar un gran exceso de un reactivo (digamos, [B]≫[A]) de forma que, a medida que progresa la reacción, sólo se consuma una pequeña fracción del reactivo en exceso (B), y pueda considerarse que su concentración permanece constante. Por ejemplo, la hidrólisis de ésteres por ácidos minerales diluidos sigue una cinética de pseudoprimer orden, en la que la concentración de agua es constante porque está presente en gran exceso:
La hidrólisis de la sacarosa (C12H22O11) en solución ácida se cita a menudo como una reacción de primer orden con velocidad
La verdadera ecuación de velocidad es de tercer orden, sin embargo, las concentraciones tanto del catalizador H+ como del disolvente H2O son normalmente constantes, por lo que la reacción es de pseudo-primer orden.[22]
Resumen para los órdenes de reacción 0, 1, 2 y n
[editar]Los pasos elementales de reacción de orden 3 (denominados reacciones ternarias) son raros y poco probables. Sin embargo, las reacciones globales compuestas de varios pasos elementales pueden, por supuesto, ser de cualquier orden (incluso no entero).
| Orden cero | Primer orden | Segundo orden | n-ésimo orden
(g = 1-n) | |
|---|---|---|---|---|
| Ley de velocidad | [23] | |||
| Ley de velocidad integrada | [23] |
[Excepto primer orden] | ||
| Unidades de la constante de velocidad (k) | ||||
| Gráfico lineal para determinar k | [A] vs. t | vs. t | vs. t | vs. t
[Excepto primer ordenr] |
| Semivida | [23] |
[Es necesario un límite para el primer orden] |
Aquí representa la concentración en molaridad (mol · L-1), es el tiempo en segundos, y la constante de velocidad de reacción. La semivida de una reacción de primer orden se expresa a menudo como t1/2 = 0,693/k (ya que ln(2)≈0,693).
Orden fraccionario
[editar]En las reacciones de orden fraccionario el orden no es un número entero, lo que a menudo indica una reacción química en cadena u otro mecanismo de reacción complejo. Por ejemplo, la pirólisis del acetaldehído (CH
3CHO ) en metano y monóxido de carbono tiene lugar con un orden de 1,5 con respecto al acetaldehído: [24] La descomposición del fosgeno (COCl
2 ) en monóxido de carbono y cloro es de orden 1 respecto al propio fosgeno y de orden 0,5 con respecto al cloro: [25]
El orden de una reacción en cadena puede racionalizarse utilizando la aproximación del estado estacionario para la concentración de intermediarios reactivos como los radicales libres. Para la pirólisis del acetaldehído, el mecanismo de Rice-Herzfeld es
- Inicio
- Propagación
- Final
donde • denota un radical libre.[24][26] Para simplificar la teoría se ignoran las reacciones del *CHO para formar un segundo *CH3.
En estado estacionario, las velocidades de formación y destrucción de radicales metilo son iguales, de modo que
de modo que la concentración del radical metilo satisfaga
La velocidad de reacción es igual a la velocidad de los pasos de propagación que forman los principales productos de reacción CH4 y CO:
de acuerdo con el orden experimental de 3/2.[24][26]
Leyes complejas
[editar]Orden mixto
[editar]Las leyes de velocidad más complejas se han descrito como de orden mixto si se aproximan a las leyes de más de un orden a diferentes concentraciones de las especies químicas implicadas. Por ejemplo, una ley de velocidad de la forma
representa reacciones concurrentes de primer orden y segundo orden (o más a menudo reacciones concurrentes de pseudo-primer orden y segundo orden), y puede describirse como mixta de primer y segundo orden.[27] Para valores suficientemente grandes de [A] una reacción de este tipo se aproximará a la cinética de segundo orden, pero para valores más pequeños de [A] la cinética se aproximará al primer orden (o pseudo-primer orden). A medida que avanza la reacción, ésta puede cambiar de segundo orden a primer orden a medida que se consume el reactivo.
Otro tipo de ley de velocidad de orden mixto tiene un denominador de dos o más términos, a menudo porque la identidad del paso que determina la velocidad depende de los valores de las concentraciones. Un ejemplo es la oxidación de un alcohol a una cetona mediante el ion hexacianoferrato (III) [Fe(CN) 6 3 − ] con el ion rutenato (VI) (RuO 4 2− ) como catalizador .[28] Para esta reacción, la velocidad de desaparición del hexacianoferrato (III) es
Es de orden cero con respecto al hexacianoferrato (III) al inicio de la reacción (cuando su concentración es alta y el catalizador de rutenio se regenera rápidamente), pero cambia a primer orden cuando su concentración disminuye y la regeneración del catalizador pasa a ser determinante de la velocidad.
Los mecanismos notables con leyes de velocidad de orden mixto con denominadores de dos términos incluyen:
- la cinética de Michaelis-Menten para la catálisis enzimática: primer orden en el sustrato (segundo orden global) a bajas concentraciones de sustrato, orden cero en el sustrato (primer orden global) a mayores concentraciones de sustrato; y
- el mecanismo de Lindemann para reacciones unimoleculares: segundo orden a bajas presiones, primer orden a altas presiones.
Orden negativo
[editar]Una velocidad de reacción puede tener un orden parcial negativo con respecto a una sustancia. Por ejemplo, la conversión de ozono (O3) en oxígeno sigue la ecuación de velocidad en un exceso de oxígeno. Esto corresponde a un segundo orden en el ozono y a un orden (-1) con respecto al oxígeno.[29]
Cuando un orden parcial es negativo el orden global suele considerarse indefinido. En el ejemplo anterior, por ejemplo, la reacción no se describe como de primer orden aunque la suma de los órdenes parciales sea , porque la ecuación de velocidad es más compleja que la de una reacción simple de primer orden.
Reacciones contrarias
[editar]Un par de reacciones directa e inversa pueden producirse simultáneamente con velocidades comparables. Por ejemplo, A y B reaccionan dando productos P y Q y viceversa (a, b, p y q son los coeficientes estequiométricos):
La expresión de la velocidad de reacción para las reacciones anteriores (suponiendo que cada una es elemental) puede escribirse como:
donde: k1 es el coeficiente de velocidad de la reacción que consume A y B; k-1 es el coeficiente de velocidad de la reacción inversa, que consume P y Q y produce A y B.
Las constantes k1 y k-1 están relacionadas con el coeficiente de equilibrio para la reacción (K) de siguiente forma (al alcanzar el equilibrio v=0):
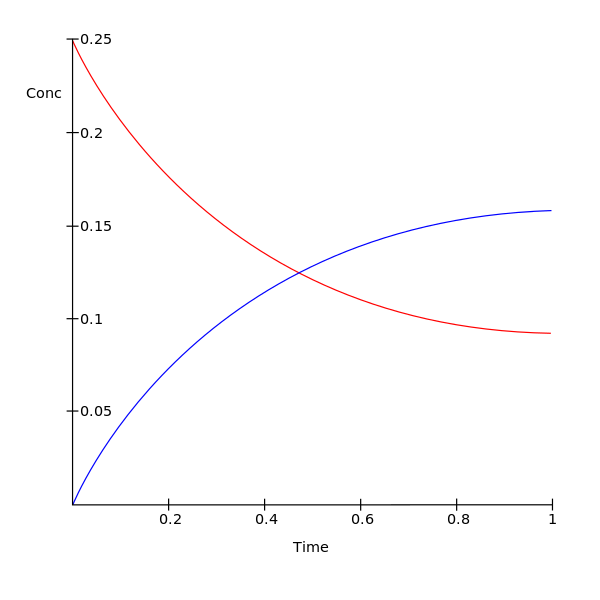
Ejemplo simple
[editar]En un equilibrio simple entre dos especies:
donde la reacción comienza con una concentración inicial del reactivo A, , y una concentración inicial de 0 para el producto P en el instante t=0.
Entonces la constante de equilibrio K se expresa como:
donde y son las concentraciones de A y P en equilibrio, respectivamente.
La concentración de A en el instante t, , está relacionada con la concentración of P en el instante t, , por la ecuación de equilibrio de la reacción:
El término no aparece porque en este ejemplo sencillo la concentración inicial de P es 0.
Esto se aplica incluso cuando el tiempo t es infinito, es decir, se ha alcanzado el equilibrio:
entonces se deduce, por la definición de K, que
y, por lo tanto,
Estas ecuaciones nos permiten desacoplar el sistema de ecuaciones diferenciales, y nos permiten resolver sólo para la concentración de A.
La ecuación de reacción se dio anteriormente como:
Para
La derivada es negativa porque esta es la velocidad de la reacción que va de A a P, y por lo tanto la concentración de A está disminuyendo. Para simplificar la notación, x es , la concentración de A en el instante t. Sea la concentración de A en el equilibrio. Entonces:
Y, por lo tanto:
la velocidad de reacción se convierte en:
lo cual da como resultado:
- .
La gráfica del logaritmo natural negativo de la concentración de A en cualquier instante menos la concentración en el equilibrio frente al tiempo da una línea recta con pendiente k1 + k-1. Mediante la medición de y se conocerán los valores de K y de las dos constantes de velocidad de reacción.[30]
Generalización del ejemplo simple
[editar]Si la concentración en el instante t = 0 es diferente de la anterior, las simplificaciones anteriores no son válidas y hay que resolver un sistema de ecuaciones diferenciales. Sin embargo, este sistema también puede resolverse exactamente para obtener las siguientes expresiones generalizadas:
Cuando la constante de equilibrio es cercana a la unidad y las velocidades de reacción muy rápidas, por ejemplo en el análisis conformacional de moléculas, se requieren otros métodos para la determinación de las constantes de velocidad, por ejemplo mediante el análisis completo de la forma de las líneas en la espectroscopia de RMN.
Reacciones consecutivas
[editar]Si las constantes de velocidad para la siguiente reacción son y ; , entonces la ecuación de la velocidad es:
- Para el reactivo A:
- Para el reactivo B:
- Para el producto C:
Con las concentraciones individuales escaladas por la población total de reactivos para convertirse en probabilidades, los sistemas lineales de ecuaciones diferenciales como estos pueden formularse como una ecuación maestra. Las ecuaciones diferenciales pueden resolverse analíticamente y las ecuaciones de velocidad integradas son
La aproximación del estado estacionario conduce a resultados muy similares de forma más sencilla.
Reacciones paralelas o competitivas
[editar]
Cuando una sustancia reacciona simultáneamente para dar dos productos diferentes, se dice que se produce una reacción paralela o competitiva.
Dos reacciones de primer orden
[editar]y , con constantes y y ecuaciones de velocidad ; y
Las ecuaciones de velocidad integradas son entonces ; y
.
Una relación importante en este caso es
Una reacción de primer orden y otra de segundo orden
[editar]Este puede ser el caso cuando se estudia una reacción bimolecular y se produce simultáneamente una hidrólisis (que puede tratarse como de pseudoorden uno): la hidrólisis complica el estudio de la cinética de la reacción, porque algún reactante se está "gastando" en una reacción paralela. Por ejemplo, A reacciona con R para dar nuestro producto C, pero mientras tanto la reacción de hidrólisis se lleva una cantidad de A para dar B, un subproducto: y .
Las ecuaciones de la velocidad son: y , donde es la pseudo constante de primer orden.[31]
La ecuación de velocidad integrada para el producto principal [C] es
, que equivale a.
La concentración de B está relacionada con la de C mediante
Las ecuaciones integradas se obtuvieron analíticamente, pero durante el proceso se supuso que . Por lo tanto, la ecuación anterior para [C] sólo puede utilizarse para concentraciones bajas de [C] en comparación con [A]0
Redes de reacciones estequiométricas
[editar]La descripción más general de una red de reacciones químicas considera un número de especies químicas distintas que reaccionan mediante reacciones.[32][33] La ecuación química de la -ésima reacción puede escribirse en la forma genérica
que suele escribirse de forma equivalente
Donde
- es el índice de reacción que va de 1 a ,
- denota la -ésima especie química,
- es la constante de velocidad de la -ésima reacción y
- y son los coeficientes estequiométricos de reactivos y productos, respectivamente.
La velocidad de una reacción de este tipo puede deducirse mediante la ley de acción de masas
que denota el flujo de moléculas por unidad de tiempo y unidad de volumen. Aquí es el vector de concentraciones. Esta definición incluye las reacciones elementales:
- reacciones de orden cero
- para las cuales para todo ,
- reacciones de primer orden
- para las cuales para un único ,
- reacciones de segundo orden
- para las cuales para exactamente dos ; es decir, una reacción bimolecular, o para un único ; es decir, una reacción de dimerización.
Cada uno de estos se analiza en detalle a continuación. Se puede definir la matriz estequiométrica.
que denota la extensión neta de moléculas de en la reacción . Las ecuaciones de velocidad de reacción pueden entonces escribirse en la forma general
Este es el producto de la matriz estequiométrica y el vector de funciones de velocidad de reacción. Existen soluciones simples particulares en equilibrio,, para sistemas compuestos por reacciones meramente reversibles. En este caso, la velocidad de las reacciones hacia delante y hacia atrás son iguales, un principio denominado equilibrio detallado. El equilibrio detallado es una propiedad de la matriz estequiométrica y no depende de la forma particular de las funciones de tipo . Todos los demás casos en los que se viola el equilibrio detallado suelen estudiarse mediante el análisis del equilibrio de flujos, que se ha desarrollado para comprender las rutas metabólicas.[34][35]
Dinámica general de la conversión unimolecular
[editar]Para una reacción unimolecular general que implica interconversión de especies diferentes, cuyas concentraciones en el instante se denotan desde hasta , es posible hallar una forma analítica que determine su evolución temporal. Dadas las constantes de velocidad de conversión de las especies a las especies denotadas como , se construye una matriz de constantes de velocidad cuyas componentes son las .
También, sea el vector de concentraciones en función del tiempo.
Sea el vector de unos.
Sea la matriz identidad .
Sea la función que toma un vector y a partir de él construye una matriz diagonal cuyas componentes de la diagonal principal son las del vector.
Sea la transformada inversa de Laplace de a .
Entonces está dado por
proporcionando así la relación entre las condiciones iniciales del sistema y su estado en el momento .
Véase también
[editar]- Cinética de Michaelis-Menten
- Molecularidad
- Matriz de petersen
- Sistema de reacción-difusión
- Reacciones en superficies: ecuaciones de velocidad para reacciones en las que al menos uno de los reactivos se adsorbe en una superficie.
- Análisis cinético del progreso de la reacción.
- Tasa de reacción
- Constante de velocidad de reacción
- Aproximación al estado estacionario
- Algoritmo de Gillespie
- Ecuación de equilibrio
- Reacción de Belousov-Zhabotinsky
- Ecuaciones de Lotka-Volterra
- Cinética química
Referencias
[editar]- ↑ Gold, Victor, ed. (2019). The IUPAC Compendium of Chemical Terminology: The Gold Book (en inglés) (4 edición). Research Triangle Park, NC: International Union of Pure and Applied Chemistry (IUPAC). doi:10.1351/goldbook.
- ↑ a b «14.3: Effect of Concentration on Reaction Rates: The Rate Law». Chemistry LibreTexts (en inglés). 18 de enero de 2015. Consultado el 10 de abril de 2023.
- ↑ Atkins y de Paula, 2006, p. 794
- ↑ Atkins y de Paula, 2006, p. 795
- ↑ a b c Atkins y de Paula, 2006, p. 796
- ↑ Connors, 1990, p. 13
- ↑ Connors, 1990, p. 12
- ↑ Atkins y de Paula, 2006, pp. 797–8
- ↑ Espenson, 1987, pp. 5–8
- ↑ Atkins y de Paula, 2006, pp. 798–800
- ↑ Espenson, 1987, pp. 15–18
- ↑ Espenson, 1987, pp. 30–31
- ↑ a b Atkins y de Paula, 2006, p. 796
- ↑ Tinoco y Wang, 1995, p. 331
- ↑ Walsh, Dylan J.; Lau, Sii Hong; Hyatt, Michael G.; Guironnet, Damien (25 de septiembre de 2017). «Kinetic Study of Living Ring-Opening Metathesis Polymerization with Third-Generation Grubbs Catalysts». Journal of the American Chemical Society (en inglés) 139 (39): 13644-13647. ISSN 0002-7863. PMID 28944665. doi:10.1021/jacs.7b08010.
- ↑ Espenson, James H. (1981). Chemical Kinetics and Reaction Mechanisms. McGraw-Hill. p. 14. ISBN 0-07-019667-2.
- ↑ Atkins y de Paula, 2006, pp. 813–4
- ↑ Keith J. Laidler, Chemical Kinetics (3rd ed., Harper & Row 1987), p.303-5 ISBN 0-06-043862-2
- ↑ R.H. Petrucci, W.S. Harwood and F.G. Herring, General Chemistry (8th ed., Prentice-Hall 2002) p.588 ISBN 0-13-014329-4
- ↑ a b Connors, 1990
- ↑ Whitten K. W., Galley K. D. and Davis R. E. General Chemistry (4th edition, Saunders 1992), pp. 638–9 ISBN 0-03-072373-6
- ↑ Tinoco y Wang, 1995, pp. 328–9
- ↑ a b c NDRL Radiation Chemistry Data Center. See also: Capellos, Christos; Bielski, Benon H. (1972). Kinetic systems: mathematical description of chemical kinetics in solution. New York: Wiley-Interscience. ISBN 978-0471134503. OCLC 247275.
- ↑ a b c Atkins y de Paula, 2006, p. 830
- ↑ Laidler, 1987, p. 301
- ↑ a b Laidler, 1987, pp. 310–311
- ↑ Espenson, 1987, pp. 34,60
- ↑ Mucientes, Antonio E.; de la Peña, María A. (November 2006). «Ruthenium(VI)-Catalyzed Oxidation of Alcohols by Hexacyanoferrate(III): An Example of Mixed Order». Journal of Chemical Education (en inglés) 83 (11): 1643. ISSN 0021-9584. doi:10.1021/ed083p1643.
- ↑ Laidler, 1987, p. 305
- ↑ Rushton, Gregory T.; Burns, William G.; Lavin, Judi M.; Chong, Yong S.; Pellechia, Perry; Shimizu, Ken D. (September 2007). «Determination of the Rotational Barrier for Kinetically Stable Conformational Isomers via NMR and 2D TLC». Journal of Chemical Education (en inglés) 84 (9): 1499. ISSN 0021-9584. doi:10.1021/ed084p1499.
- ↑ Manso, José A.; Pérez-Prior, M. Teresa; García-Santos, M. del Pilar; Calle, Emilio; Casado, Julio (2005). «A Kinetic Approach to the Alkylating Potential of Carcinogenic Lactones». Chemical Research in Toxicology 18 (7): 1161-1166. PMID 16022509. doi:10.1021/tx050031d.
- ↑ Heinrich, Reinhart; Schuster, Stefan (2012). The Regulation of Cellular Systems. Springer Science & Business Media. ISBN 9781461311614.
- ↑ Chen, Luonan; Wang, Ruiqi; Li, Chunguang; Aihara, Kazuyuki (2010). Modeling Biomolecular Networks in Cells. ISBN 978-1-84996-213-1. doi:10.1007/978-1-84996-214-8.
- ↑ Szallasi, Z., and Stelling, J. and Periwal, V. (2006) System modeling in cell biology: from concepts to nuts and bolts. MIT Press Cambridge.
- ↑ Iglesias, Pablo A.; Ingalls, Brian P. (2010). Control theory and systems biology. MIT Press. ISBN 9780262013345.
Bibliografía
[editar]- Atkins, Peter; de Paula, Julio (2006). «The rates of chemical reactions». Atkins' Physical chemistry (8th edición). W.H. Freeman. pp. 791–823. ISBN 0-7167-8759-8.
- Connors, Kenneth Antonio (1990). Chemical kinetics : the study of reaction rates in solution. John Wiley & Sons. ISBN 9781560810063.
- Espenson, James H. (1987). Chemical kinetics and reaction mechanisms (2nd edición). McGraw Hill. ISBN 9780071139496.
- Laidler, Keith James (1987). Chemical kinetics (3rd edición). Harper & Row. ISBN 9780060438623.
- Tinoco, Ignacio Jr.; Wang, James C. (1995). Physical chemistry : principles and applications in biological sciences (3rd edición). Prentice Hall. ISBN 9780131865457.
Enlaces externos
[editar]- Cinética química, velocidad de reacción y orden (necesita flash player)
- Cinética de reacción, ejemplos de leyes de velocidad importantes (conferencia con audio).
- Velocidades de reacción
| Control de autoridades |
|
|---|
Datos: Q904041