Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness.[1] In DM, muscles are often unable to relax after contraction.[1] Other manifestations may include cataracts, intellectual disability and heart conduction problems.[1][2] In men, there may be early balding and infertility.[1] While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.[1]
| Myotonic dystrophy | |
|---|---|
| Other names | Dystrophia myotonica,[1] myotonia atrophica,[1] myotonia dystrophica,[1] Curschmann–Batten–Steinert syndrome |
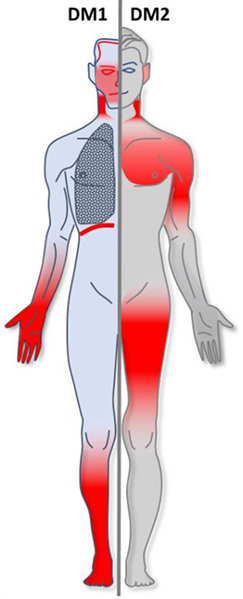 | |
| Areas of body affected in myotonic dystrophy, types 1 and 2, colored in red | |
| Specialty | Neurology, neuromuscular medicine, physical medicine and rehabilitation, medical genetics, pediatrics |
| Symptoms | Muscle loss, weakness, muscles which myotonia[1] |
| Complications | Cataracts, intellectual disability, heart conduction problems[1][2] |
| Usual onset | 20s to 30s[1] |
| Duration | Long term[1] |
| Types | Type 1, type 2[1] |
| Causes | Genetic disorder (autosomal dominant)[1] |
| Diagnostic method | Genetic testing.[2] |
| Treatment | Braces, wheelchair, pacemakers, non invasive positive pressure ventilation[2] |
| Medication | Mexiletine, carbamazepine, tricyclic antidepressants, nonsteroidal anti inflammatory drugs[2] |
| Frequency | >1 in 8,000 people[1] |
Myotonic dystrophy is caused by a genetic mutation in one of two genes. Mutation of the DMPK gene causes myotonic dystrophy type 1 (DM1). Mutation of CNBP gene causes type 2 (DM2).[1] DM is typically inherited, following an autosomal dominant inheritance pattern,[1] and it generally worsens with each generation.[1] A type of DM1 may be apparent at birth.[1] DM2 is generally milder.[1] Diagnosis is confirmed by genetic testing.[2]
There is no cure.[3] Treatments may include braces or wheelchairs, pacemakers and non-invasive positive pressure ventilation.[2] The medications mexiletine or carbamazepine can help relax muscles.[2] Pain, if it occurs, may be treated with tricyclic antidepressants and nonsteroidal anti-inflammatory drugs (NSAIDs).[2]
Myotonic dystrophy affects about 1 in 2,100 people,[4] a number that was long estimated to be much lower (often cited as 1 in 8,000), reflecting that not all patients have immediate symptoms and, once they do have symptoms, the long time it typically takes to get to the right diagnosis.[1] It is the most common form of muscular dystrophy that begins in adulthood.[1] It was first described in 1909, with the underlying cause of type 1 determined in 1992.[2]
Signs and symptoms
editDM causes muscle weakness, early onset of cataracts, and myotonia, which is delayed relaxation of muscles after contraction.[5] Cataracts can be either a cortical cataract with a blue dot appearance, or a posterior subcapsular cataract.[6] Other organs affected include the heart, lungs, gastrointestinal tract, skin, and brain.[5] Insulin resistance can also occur.[5] Signs and symptoms vary considerably by severity, unusual phenotype, and form (DM1/DM2).[citation needed] DM1 and DM2 differ in regards to the muscles they affect, age of onset, severity of disease, and extramuscular manifestations.[5]
DM1
editDM1 usually begins in the muscles of the hands, feet, neck, or face.[5] One manifestation of facial weakness is drooping of the eyelid (ptosis).[citation needed] It slowly progresses to involve other muscle groups, including the heart. Myotonia tends to be more prominent in DM1 compared to DM2.[5] Other DM1 manifestations include problems with executive function (e.g., organization, concentration, word-finding) and hypersomnia.[5] Abnormalities in the electrical activity of the heart are common in DM1, manifesting as arrhythmias or conduction blocks.[2] Sometimes, dilated cardiomyopathy occurs.[2] Symptoms onset any time from birth to adulthood.[5] The earlier the disease onset, the greater the variety of possible signs and symptoms. [citation needed] Thus, various diagnostic classifications based on the age of onset/severity of the disease have been proposed, although DM1 manifestations likely lie on a continuum.[7]
Congenital DM1
editWhen DM1 onsets at birth, it is called congenital DM1.[5] Manifestations that can be present at birth include hypotonia, respiratory failure, feeding difficulty, and club foot (talipes equinovarus), any of which tend to resolve over several years.[5] During childhood, intellectual impairment, attention deficit hyperactivity disorder (ADHD), and autism spectrum disorders (ASD) can result.[5] Gastrointestinal issues can result, which can be severe, manifestations including diarrhea, constipation, and fecal incontinence.[5] The symptoms of adult DM often manifest during adolescence.[5] Infantile DM1 can be distinguished as another disease category, or it can be grouped with congenital DM1 or childhood-onset DM1.[7][citation needed]
Childhood-onset DM1
editChildhood-onset DM1 is defined as onset of symptoms between ages 1 and 10 years.[5] Manifestations include the same intellectual and gastrointestinal symptoms seen in congenital DM1.[5]
DM2
editDM2 is generally milder than DM1, with generally fewer DM2 people requiring assistive devices than DM1 people.[8] DM2 preferentially affects muscles closer to or on the torso, including the neck flexors, hip flexors, and hip extensors.[5] Muscle pain is prominent in DM2.[5] Heart issues, while still potentially fatal, are less common and severe in DM2 than DM1.[2] Symptoms onset in early to late adulthood.[5] Severe congenital onset, which can occur in DM1, has not been observed in DM2.[8]
Genetics
editMyotonic dystrophy (DM) is a genetic condition that is inherited in an autosomal dominant pattern, meaning each child of an affected individual has a 50% chance of inheriting the disease. The mutation involves satellite DNA, which is tandemly repeated sequences of DNA that do not code for a protein. The repeats implicated in myotonic dystrophy are either 3 or 4 nucleotides in length, classified as microsatellites. Disease results from an abnormally increased number of these microsatellites, termed microsatellite expansion.
DM1
editThe microsatellite expansion responsible for DM1 is of cytosine-thymine-guanine (CTG) triplet repeats, termed trinucleotide repeat expansion and classifying DM1 as one of several trinucleotide repeat disorders. This expansion occurs at the end of the DMPK gene, in the 3' untranslated region. DMPK is located on the long arm of chromosome 19.[9][10] DMPK codes for myotonic dystrophy protein kinase,[11] a protein expressed predominantly in skeletal muscle.[12]
Between 5 and 37 repeats is considered normal; between 38 and 49 repeats is considered pre-mutation, and although not producing symptoms, children can have further repeat expansion and symptomatic disease;[13] greater than 50 repeats almost invariably is symptomatic, with some noted exceptions. Longer repeats are usually associated with earlier onset and more severe disease.[14]
DMPK alleles with greater than 37 repeats are unstable and additional trinucleotide repeats may be inserted during cell division in mitosis and meiosis. Consequently, the children of individuals with premutations or mutations inherit DMPK alleles which are longer than their parents and therefore are more likely to be affected or display an earlier onset and greater severity of the condition, a phenomenon known as anticipation. Repeat expansion is generally considered to be a consequence of the incorporation of additional bases as a result of strand slippage during either DNA replication or DNA repair synthesis.[15] Misalignments occurring during homologous recombinational repair, double-strand break repair or during other DNA repair processes likely contribute to trinucleotide repeat expansions in DM1.[15] Paternal transmission of the congenital form is uncommon (13%), possibly due to selection pressures against sperm with expanded repeats, but juvenile or adult-onset is equally transmitted from either parent. Anticipation tends to be less severe than in cases of maternal inheritance.[citation needed]
The RNA from the expanded trinucleotide repeat region forms intranucleoplasmic hairpin loops due to the extensive hydrogen bonding between C-G base pairs, and it has been demonstrated that these sequester the splicing regulator MBNL1 to form distinctive foci.[16]
A severe form of DM1, congenital myotonic dystrophy, may appear in newborns of mothers who have DM. Congenital myotonic dystrophy can also be inherited via the paternal gene, although it is said to be relatively rare. Congenital means that the condition is present from birth.[8]
DM2
editThe microsatellite expansion responsible for DM2 is of cytosine-cytosine-thymine-guanine (CCTG) repeats, classifying it as a tetranucleotide repeat disorder. This expansion occurs in the first intron CNBP gene on chromosome 3.[17][18][19]
The repeat expansion for DM2 is much larger than for DM1, ranging from 75 to over 11,000 repeats.[17] Like DM1, the size of the microsatellite repeat array lengthens from generation to generation.[5] Unlike DM1, anticipation does not result, as the degree of repeat expansion beyond 75 repeats does not affect the age of onset or disease severity.[5][13]
The repeat expansion produces an RNA transcript that binds to RNA-binding proteins such as MBNL1, as in DM1.[5] Also, repeat expansion likely reduces expression of CNBP, loss of which causes muscle toxicity.[5]
Pathophysiology
editMolecular
editMutations of DM1 and DM2 cause production of RNA that sequesters RNA-binding proteins, causing dysregulated RNA splicing.[5] This dysregulated RNA splicing is particularly toxic to skeletal, cardiac, and smooth muscle.[20] One example in DM1 involves the chloride channel ClC-1.[5] Mutated DMPK RNA binds to MBNL1, causing ClC-1 pre-mRNA to be spliced into the fetal form instead of the adult form.[5] Functional loss of the chloride channel causes myotonia.[5]
Histology
editIn DM1, there can be increased central nuclei, angular fibers, fiber atrophy, and pyknotic clumps.[5] There can be selective atrophy of type 1 muscle fibers.[21] Muscle fibers show signs of degeneration and regeneration.[21] There is modest fibrosis of the endomysium.[21]
In DM2, there can be variation in the sizes of muscle fibers, although often there are no abnormalities.[5] There is selective atrophy of type 2 muscle fibers. Again, there are central nuclei and nuclear clumps.[21]
Diagnosis
editThis section needs more reliable medical references for verification or relies too heavily on primary sources. (April 2016) |
The diagnosis of DM1 and DM2 can be difficult due to the large number of neuromuscular disorders, most of which are very rare. One study found that diagnosis is made an average of seven years after symptom onset for DM1, and fourteen years for DM2.[22][5]
As a result, people with multiple symptoms that may be explained by a complex disorder such as DM1 or DM2 will generally be referred by their primary care physician to a neurologist for diagnosis. Depending on the presentation of symptoms, people may be referred to a number of medical specialists including cardiologists, ophthalmologists, endocrinologists, and rheumatologists. In addition, the clinical presentation is obscured by the degree of severity or the presence of unusual phenotypes.
Though there is presently no cure for DM and management is currently symptom-based, a precise diagnosis is still necessary to anticipate multiple other problems that may develop over time (e.g. cataracts). An accurate diagnosis is important to assist with appropriate medical monitoring and management of symptoms. In addition, genetic counseling should be made available to all people because of the high risk of transmission. Potentially serious anesthetic risks are important to note, so the presence of this disorder should be brought to the attention of all medical providers.
Classification
edit| Type | Gene | Repeat | Anticipation | Severity |
|---|---|---|---|---|
| DM1 | DMPK | CTG | Yes | Moderate-severe |
| DM2 | ZNF9 | CCTG | Minimal/none | Mild-moderate |
There are two main types of myotonic dystrophy. Type 1 (DM1), also known as Steinert disease, has a severe congenital form and a milder childhood-onset form as well as an adult-onset form.[23] This disease is most often in the facial muscles, levator palpebrae superioris, temporalis, sternocleidomastoids, distal muscles of the forearm, hand intrinsic muscles, and ankle dorsiflexors.[24] Type 2 (DM2), also known as proximal myotonic myopathy (PROMM), is rarer and generally manifests with milder signs and symptoms than DM1.[8]
Other forms of myotonic dystrophy not associated with DM1 or DM2 genetic mutations have been described.[13] One case which was proposed as a candidate for the "DM3" label,[25] was later characterized as an unusual form of inclusion body myopathy associated with Paget's disease and frontotemporal dementia.[13][18][26]
Genetic testing
editGenetic tests, including prenatal testing, are available for both confirmed forms. Molecular testing is considered the gold standard of diagnosis.
Prenatal testing
editTesting at pregnancy to determine whether an unborn child is affected is possible if genetic testing in a family has identified a DMPK mutation. This can be done at 10–12 weeks gestation by a procedure called chorionic villus sampling (CVS) that involves removing a tiny piece of the placenta and analyzing DNA from its cells. It can also be done by amniocentesis after 14 weeks gestation by removing a small amount of the amniotic fluid surrounding the baby and analyzing the cells in the fluid. Each of these procedures has a small risk of miscarriage associated with it and those who are interested in learning more should check with their doctor or genetic counselor. There is also another procedure called preimplantation diagnosis that allows a couple to have a child that is unaffected by the genetic condition in their family. This procedure is experimental and not widely available. Those interested in learning more about this procedure should check with their doctor or genetic counselor.
Predictive testing
editIt is possible to test someone who is at risk for developing DM1 before they are showing symptoms to see whether they inherited an expanded trinucleotide repeat. This is called predictive testing. Predictive testing cannot determine the age of onset that someone will begin to have symptoms or the course of the disease. If the child is not having symptoms, the testing is not possible with an exception of emancipated minors as a policy.
Auxiliary testing
editElectrodiagnostic testing (EMG and NCS) can detect the electrical signs of myotonia before myotonia becomes noticeable to the affected individual.[5]
Muscle biopsy can reveal damage of the muscle, but findings are generally nonspecific and do not greatly aid in diagnosis.[5]
Management
editThere is currently no cure for or treatment specific to myotonic dystrophy. Management is focused on the complications of the disease, particularly those related to the lungs and heart, which are life-threatening.[27] Complications relating to the cardiopulmonary system account for 70% of deaths due to DM1.[13] Compromised lung function can, in turn, contribute to life-threatening complications during anesthesia and pregnancy.[27]
Lung complications are the leading cause of death in DM1, warranting lung function monitoring with pulmonary function tests every 6 months.[27] Central sleep apnea or obstructive sleep apnea may cause excessive daytime sleepiness, and these individuals should undergo a sleep study. Non-invasive ventilation may be offered if there is an abnormality. Otherwise, there is evidence for the use of modafinil as a central nervous system stimulant, although a Cochrane review has described the evidence thus far as inconclusive.[citation needed]
Cardiac complications are the second leading cause of death in DM1, and commonly no symptoms are present prior to adverse events.[27] All affected individuals are advised to have an annual or biennial ECG.[27] Pacemaker insertion may be required for individuals with cardiac conduction abnormalities. Improving the quality of life which can be measured using specific questionnaires[28] is also a main objective of the medical care.
Physical activity
editThere is a lack of high-quality evidence to determine the effectiveness and the safety of physical activities for people who have myotonic dystrophy.[29] Further research is required to determine if combined strength and aerobic training at moderate intensity is safe for people who have neuromuscular diseases, however the combination of aerobic and strength exercises may increase muscle strength.[30][29] Aerobic exercise via stationary bicycle with an ergometer may be safe and effective in improving fitness in people with DM1.[31] Cardiovascular impairments and myotonic sensitivities to exercise and temperature necessitate close monitoring of people and educating people in self-monitoring during exercise via the Borg scale, heart rate monitors, and other physical exertion measurements.[32]
Orthotics
editMuscular weakness of dorsiflexors (dorsiflexion) hinders the ability to clear the floor during the swing phase of gait and people may adopt a steppage gait pattern[32] or ankle-foot-orthotics may be indicated.[13] Factors such as hand function, skin integrity, and comfort must be assessed prior to prescription. Neck braces can also be prescribed for neck muscle weakness.[13]
Mobility aids and adaptive equipment
editUpper and lower limb weakness, visual impairments and myotonia may lead to the need for mobility aids and functional adaptive equipment such as buttonhooks and handled sponges for optimal hand function. If assistive devices and home adaptations are needed, physical therapists may refer on to occupational therapist(s) for further assessment.[13]
Prognosis
editLife expectancy in non-congenital late-onset or adult onset DM1 is in the early 50s,[5] with pulmonary complications being the leading cause of death, followed by cardiac complications.[27] DM2 life expectancy has yet to be studied.[5]
Epidemiology
editThe prevalence of DM1 ranges from 5 to 20 per 100,000 (1:20,000–1:5000).[5] Up to 48 per 100,000 (1:2100) of individuals tested positive for the mutation of DM1 in New York, although not all of these individuals would have become symptomatic.[33] Again in New York, premutations for DM1 were found in 191 per 100,000 (1:525).[33] DM2 prevalence is not known, but genetic studies estimate it to be as high as 1:1830.[5] DM affects males and females approximately equally.[citation needed] About 30,000 people in the United States are affected.[citation needed] In most populations, DM1 appears to be more common than DM2. However, recent studies suggest that type 2 may be as common as type 1 among people in Germany and Finland.[1]
DM1 is the most common form of myotonic muscular dystrophy diagnosed in children, with a prevalence ranging from 1 per 100,000 in Japan to 3–15 per 100,000 in Europe.[13] The prevalence may be as high as 1 in 500 in regions such as Quebec, possibly due to the founder effect. The incidence of congenital myotonic dystrophy is thought to be about 1:20,000.
History
editMyotonic dystrophy was first described by a German physician, Hans Gustav Wilhelm Steinert, who first published a series of six cases of the condition in 1909.[34] Isolated case reports of myotonia had been published previously, including reports by Frederick Eustace Batten and Hans Curschmann, and type 1 myotonic dystrophy is therefore sometimes known as Curschmann-Batten-Steinert syndrome.[35] The underlying cause of type 1 myotonic dystrophy was determined in 1992.[2]
Research directions
editAltered splicing of the muscle-specific chloride channel 1 (ClC-1) has been shown to cause the myotonic phenotype of DM1 and is reversible in mouse models using Morpholino antisense to modify splicing of ClC-1 mRNA.[36]
Some small studies have suggested that imipramine, clomipramine and taurine may be useful in the treatment of myotonia.[13] However, due to the weak evidence and potential side effects such as cardiac arrhythmias, these treatments are rarely used. A recent study in December 2015 showed that a common FDA approved antibiotic, erythromycin, reduced myotonia in mice.[37] Human studies are planned for erythromycin. Erythromycin has been used successfully in patients with gastric issues.[38]
References
edit- ^ a b c d e f g h i j k l m n o p q r s t u v w "myotonic dystrophy". GHR. 11 October 2016. Archived from the original on 18 October 2016. Retrieved 16 October 2016.
- ^ a b c d e f g h i j k l m n Meola, G; Cardani, R (April 2015). "Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1852 (4): 594–606. doi:10.1016/j.bbadis.2014.05.019. PMID 24882752.
- ^ Klein, AF; Dastidar, S; Furling, D; Chuah, MK (2015). "Therapeutic Approaches for Dominant Muscle Diseases: Highlight on Myotonic Dystrophy". Current Gene Therapy. 15 (4): 329–37. doi:10.2174/1566523215666150630120537. PMID 26122101.
- ^ Johnson, N. (2021). "Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program". Neurology. 96 (7): e1045–e1053. doi:10.1212/WNL.0000000000011425. PMC 8055332. PMID 33472919.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai Johnson, NE (December 2019). "Myotonic Muscular Dystrophies". Continuum (Minneapolis, Minn.). 25 (6): 1682–1695. doi:10.1212/CON.0000000000000793. PMID 31794466. S2CID 208531759.
- ^ Yanoff, Myron; Jay S. Duker (2008). Ophthalmology (3rd ed.). Edinburgh: Mosby. p. 411. ISBN 978-0323057516.
- ^ a b Ho, G; Carey, KA; Cardamone, M; Farrar, MA (January 2019). "Myotonic dystrophy type 1: clinical manifestations in children and adolescents". Archives of Disease in Childhood. 104 (1): 48–52. doi:10.1136/archdischild-2018-314837. hdl:1959.4/unsworks_61891. PMID 29871899. S2CID 46949763.
- ^ a b c d "Myotonic Dystrophies". The Lecturio Medical Concept Library. Retrieved 11 August 2021.
- ^ Harley HG, Walsh KV, Rundle S, Brook JD, Sarfarazi M, Koch MC, Floyd JL, Harper PS, Shaw DJ (May 1991). "Localisation of the myotonic dystrophy locus to 19q13.2-19q13.3 and its relationship to twelve polymorphic loci on 19q". Hum. Genet. 87 (1): 73–80. doi:10.1007/BF01213096. PMID 2037285. S2CID 31229908.
- ^ Bird, Thomas D. (1 January 1993). "Myotonic Dystrophy Type 1". GeneReviews. PMID 20301344. Archived from the original on 18 January 2017. Retrieved 9 May 2016.update 2015
- ^ Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O'Hoy K (March 1992). "Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene". Science. 255 (5049): 1253–5. Bibcode:1992Sci...255.1253M. doi:10.1126/science.1546325. PMID 1546325.
- ^ van der Ven PF, Jansen G, van Kuppevelt TH, Perryman MB, Lupa M, Dunne PW, ter Laak HJ, Jap PH, Veerkamp JH, Epstein HF (November 1993). "Myotonic dystrophy kinase is a component of neuromuscular junctions". Hum. Mol. Genet. 2 (11): 1889–94. doi:10.1093/hmg/2.11.1889. PMID 8281152.
- ^ a b c d e f g h i j Turner C, Hilton-Jones D (April 2010). "The myotonic dystrophies: diagnosis and management". J. Neurol. Neurosurg. Psychiatry. 81 (4): 358–67. doi:10.1136/jnnp.2008.158261. PMID 20176601.
- ^ Yum, Kevin; Wang, Eric T; Kalsotra, Auinash (2017). "Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes". Current Opinion in Genetics & Development. 44: 30–37. doi:10.1016/j.gde.2017.01.007. ISSN 0959-437X. PMC 5447468. PMID 28213156.
- ^ a b Usdin K, House NC, Freudenreich CH (2015). "Repeat instability during DNA repair: Insights from model systems". Crit. Rev. Biochem. Mol. Biol. 50 (2): 142–67. doi:10.3109/10409238.2014.999192. PMC 4454471. PMID 25608779.
- ^ Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA (July 2005). "Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy". J. Cell Sci. 118 (Pt 13): 2923–33. doi:10.1242/jcs.02404. PMID 15961406.
- ^ a b Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP (February 2003). "Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum". Neurology. 60 (4): 657–64. doi:10.1212/01.wnl.0000054481.84978.f9. PMID 12601109. S2CID 35860531.
- ^ a b Dalton, Joline C.; Ranum, Laura P.W.; Day, John W. (1 January 1993). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Myotonic Dystrophy Type 2. Seattle (WA): University of Washington, Seattle. PMID 20301639. Archived from the original on 28 January 2017.updated 2013
- ^ Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP (August 2001). "Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9". Science. 293 (5531): 864–7. doi:10.1126/science.1062125. PMID 11486088. S2CID 30903810.
- ^ Peterson, Janel A. M.; Cooper, Thomas A. (26 November 2022). "Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2". International Journal of Molecular Sciences. 23 (23): 14779. doi:10.3390/ijms232314779. ISSN 1422-0067. PMC 9737721. PMID 36499107.
- ^ a b c d Gherardi, Romain; Amato, Anthony A.; Lidov, Hart G.; Girolami, Umberto De (November 2018). "Pathology of Skeletal Muscle". In Gray, Francoise; Duyckaerts, Charles; Girolami, Umberto de (eds.). Escourolle and Poirier's manual of basic neuropathology (Sixth ed.). New York, NY: Oxford University Press. doi:10.1093/med/9780190675011.001.0001. ISBN 9780190675011.
- ^ Hilbert, JE; Ashizawa, T; Day, JW; Luebbe, EA; Martens, WB; McDermott, MP; Tawil, R; Thornton, CA; Moxley RT, 3rd (October 2013). "Diagnostic odyssey of patients with myotonic dystrophy". Journal of Neurology. 260 (10): 2497–504. doi:10.1007/s00415-013-6993-0. PMC 4162528. PMID 23807151.
{{cite journal}}: CS1 maint: numeric names: authors list (link) - ^ "Juvenile-Onset MMD1". Muscular Dystrophy Association. MDA. Archived from the original on 8 March 2015. Retrieved 17 March 2015.
- ^ "Myotonic dystrophy: Etiology, clinical features, and diagnosis". Archived from the original on 22 February 2014. Retrieved 15 February 2014.
- ^ Le Ber I, Martinez M, Campion D, Laquerrière A, Bétard C, Bassez G, Girard C, Saugier-Veber P, Raux G, Sergeant N, Magnier P, Maisonobe T, Eymard B, Duyckaerts C, Delacourte A, Frebourg T, Hannequin D (September 2004). "A non-DM1, non-DM2 multisystem myotonic disorder with frontotemporal dementia: phenotype and suggestive mapping of the DM3 locus to chromosome 15q21-24". Brain. 127 (Pt 9): 1979–92. doi:10.1093/brain/awh216. PMID 15215218.
- ^ Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, Kress W, Schoser B, Moxley R (June 2006). "140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management". Neuromuscul. Disord. 16 (6): 403–13. doi:10.1016/j.nmd.2006.03.010. PMID 16684600. S2CID 37453148.
- ^ a b c d e f Ashizawa, T; Gagnon, C; Groh, WJ; Gutmann, L; Johnson, NE; Meola, G; Moxley R, 3rd; Pandya, S; Rogers, MT; Simpson, E; Angeard, N; Bassez, G; Berggren, KN; Bhakta, D; Bozzali, M; Broderick, A; Byrne, JLB; Campbell, C; Cup, E; Day, JW; De Mattia, E; Duboc, D; Duong, T; Eichinger, K; Ekstrom, AB; van Engelen, B; Esparis, B; Eymard, B; Ferschl, M; Gadalla, SM; Gallais, B; Goodglick, T; Heatwole, C; Hilbert, J; Holland, V; Kierkegaard, M; Koopman, WJ; Lane, K; Maas, D; Mankodi, A; Mathews, KD; Monckton, DG; Moser, D; Nazarian, S; Nguyen, L; Nopoulos, P; Petty, R; Phetteplace, J; Puymirat, J; Raman, S; Richer, L; Roma, E; Sampson, J; Sansone, V; Schoser, B; Sterling, L; Statland, J; Subramony, SH; Tian, C; Trujillo, C; Tomaselli, G; Turner, C; Venance, S; Verma, A; White, M; Winblad, S (December 2018). "Consensus-based care recommendations for adults with myotonic dystrophy type 1". Neurology. Clinical Practice. 8 (6): 507–520. doi:10.1212/CPJ.0000000000000531. PMC 6294540. PMID 30588381.
{{cite journal}}: CS1 maint: numeric names: authors list (link) - ^ Dany A, Barbe C, Rapin A, Réveillère C, Hardouin JB, Morrone I, Wolak-Thierry A, Dramé M, Calmus A, Sacconi S, Bassez G, Tiffreau V, Richard I, Gallais B, Prigent H, Taiar R, Jolly D, Novella JL, Boyer FC (November 2015). "Construction of a Quality of Life Questionnaire for slowly progressive neuromuscular disease". Qual Life Res. 24 (11): 2615–23. doi:10.1007/s11136-015-1013-8. PMID 26141500. S2CID 25834947.
- ^ a b Voet, Nicoline Bm; van der Kooi, Elly L.; van Engelen, Baziel Gm; Geurts, Alexander Ch (2019). "Strength training and aerobic exercise training for muscle disease". The Cochrane Database of Systematic Reviews. 2019 (12): CD003907. doi:10.1002/14651858.CD003907.pub5. ISSN 1469-493X. PMC 6953420. PMID 31808555.
- ^ Cup EH, Pieterse AJ, Ten Broek-Pastoor JM, Munneke M, van Engelen BG, Hendricks HT, van der Wilt GJ, Oostendorp RA (November 2007). "Exercise therapy and other types of physical therapy for patients with neuromuscular diseases: a systematic review". Arch Phys Med Rehabil. 88 (11): 1452–64. doi:10.1016/j.apmr.2007.07.024. PMID 17964887.
- ^ Orngreen MC, Olsen DB, Vissing J (May 2005). "Aerobic training in patients with myotonic dystrophy type 1". Ann. Neurol. 57 (5): 754–7. doi:10.1002/ana.20460. PMID 15852373. S2CID 26547411.
- ^ a b Pandya, S; Eichinger, K. "Role of physical therapy in the assessment and management of individuals with myotonic dystrophy". Myotonic Dystrophy Foundation. Archived from the original on 26 September 2015. Retrieved 5 May 2012.
- ^ a b Johnson, NE; Butterfield, RJ; Mayne, K; Newcomb, T; Imburgia, C; Dunn, D; Duval, B; Feldkamp, ML; Weiss, RB (16 February 2021). "Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program". Neurology. 96 (7): e1045–e1053. doi:10.1212/WNL.0000000000011425. PMC 8055332. PMID 33472919.
- ^ Mishra SK, Singh S, Lee B, Khosa S, Moheb N, Tandon VA (2018). ""Dystrophia Myotonica" and the Legacy of Hans Gustav Wilhelm Steinert". Ann Indian Acad Neurol. 21 (2): 116–118. doi:10.4103/aian.AIAN_182_17. PMC 6073962. PMID 30122835.
- ^ Olbrych-Karpińska B, Tutaj A (September 1981). "[Case of Curschmann-Batten-Steinert syndrome]". Wiad. Lek. (in Polish). 34 (17): 1467–9. ISSN 0043-5147. PMID 7331343.
- ^ Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA (December 2007). "Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy". J. Clin. Invest. 117 (12): 3952–7. doi:10.1172/JCI33355. PMC 2075481. PMID 18008009.
- ^ Nakamori M, Taylor K, Mochizuki H, Sobczak K, Takahashi MP (January 2016). "Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy". Annals of Clinical and Translational Neurology. 3 (1): 42–54. doi:10.1002/acn3.271. PMC 4704483. PMID 26783549.
- ^ Rönnblom A, Andersson S, Hellström PM, Danielsson A (August 2002). "Gastric emptying in myotonic dystrophy". Eur. J. Clin. Invest. 32 (8): 570–4. doi:10.1046/j.1365-2362.2002.01028.x. PMID 12190956. S2CID 12510078.