Multiple system atrophy
| Multiple system atrophy | |
|---|---|
 | |
| Alpha-synuclein immunohistochemistry of the brain showing many glial inclusion bodies | |
| Specialty | Neurology |
| Symptoms | Parkinsonism, xerostomia, dysautonomia, ataxia |
| Complications | Cardiac arrest, infections, aspiration pneumonia |
| Usual onset | 50–60 years |
| Duration | Long term |
| Types |
|
| Causes | Unknown |
| Diagnostic method | MRI, CT scan, autopsy |
| Treatment | Physical therapy, hospice care |
| Medication | L-DOPA, fludrocortisone, midodrine |
| Prognosis | Life expectancy 6–12 years after onset of symptoms |
| Frequency | 5 per 100,000 people |
Multiple system atrophy (MSA) is a rare neurodegenerative disorder[1] characterized by tremors, slow movement, muscle rigidity, postural instability (collectively known as parkinsonism), autonomic dysfunction and ataxia. This is caused by progressive degeneration of neurons in several parts of the brain including the basal ganglia, inferior olivary nucleus, and cerebellum.
Many people affected by MSA experience dysfunction of the autonomic nervous system, which commonly manifests as orthostatic hypotension, impotence, loss of sweating, dry mouth and urinary retention and incontinence. Palsy of the vocal cords is an important and sometimes initial clinical manifestation of the disorder.
A prion of the alpha-synuclein protein within affected neurons may cause MSA.[2] About 55% of MSA cases occur in men, with those affected first showing symptoms at the age of 50–60 years.[3] MSA often presents with some of the same symptoms as Parkinson's disease. However, those with MSA generally show little response to the dopamine agonists used to treat Parkinson's disease and only about 9% of MSA patients with tremor exhibit a true parkinsonian pill-rolling tremor.[4]
MSA is distinct from multisystem proteinopathy, a more common muscle-wasting syndrome. MSA is also different from multiple organ dysfunction syndrome, sometimes referred to as multiple organ failure, and from multiple organ system failures, an often-fatal complication of septic shock and other severe illnesses or injuries.
Signs and symptoms
[edit]MSA is characterized by the following: Autonomic and at least one Motor (clinically established MSA criteria 2022)[5][6]
- autonomic dysfunction: Post-void urinary residual volume ≥100 mL (usually by ultrasound); Unexplained urinary urge incontinence; or Neurogenic orthostatic hypotension (≥20/10 mmHg blood pressure drop) within 3 minutes (usually by head‐up tilt)
- parkinsonism (muscle rigidity +/ tremor and slow movement: MSA-P)
- cerebellar ataxia (Poor coordination/unsteady walking: MSA-C)
A variant with combined features of MSA and dementia with Lewy bodies may also exist.[unreliable medical source?][7] There have also been occasional instances of frontotemporal lobar degeneration associated with MSA.[8]
Initial presentation
[edit]The most common first sign of MSA is the appearance of an "akinetic-rigid syndrome" (i.e. slowness of initiation of movement resembling Parkinson's disease) found in 62% at first presentation. Other common signs at onset include problems with balance (cerebellar ataxia) found in 22% at first presentation, followed by genito-urinary symptoms (9%): both men and women often experience urgency, frequency, incomplete bladder emptying, or an inability to pass urine (retention). About 1 in 5 MSA patients experience a fall in their first year of disease.[9]
For men, the first sign can be erectile dysfunction. Women have also reported reduced genital sensitivity.[10]
Progression
[edit]As the disease progresses, one of three groups of symptoms predominates. These are:[11]
- Parkinsonism - slow, stiff movement, writing becomes small and spidery[12][13]
- Cerebellar dysfunction - difficulty coordinating movement and balance[14]
- Autonomic nervous system dysfunction - impaired automatic body functions, including one, some, or all of the following:[15]
- postural or orthostatic hypotension, resulting in dizziness or fainting upon standing up[16]
- urinary incontinence or urinary retention[17][18][19]
- impotence[20]
- constipation[21]
- vocal cord paralysis
- dry mouth and skin
- trouble regulating body temperature due to sweating deficiency in all parts of the body
- loud snoring, abnormal breathing or inspiratory stridor during sleep
- other sleep disorders including sleep apnea, REM behavior disorder[22]
- double vision[23]
- muscle twitches[23]
- Cognitive impairment[24]
Genetics
[edit]One study found a correlation between the deletion of genes in a specific genetic region and the development of MSA in a group of Japanese patients. The region in question includes the SHC2 gene which, in mice and rats, appears to have some function in the nervous system. The authors of this study hypothesized that there may be a link between the deletion of the SHC2 and the development of MSA.[25]
A follow-up study was unable to replicate this finding in American MSA patients.[26] The authors of the study concluded that "Our results indicate that SHC2 gene deletions underlie few, if any, cases of well-characterized MSA in the US population. This is in contrast to the Japanese experience reported by Sasaki et al., likely reflecting heterogeneity of the disease in different genetic backgrounds."[clarification needed]
Another study investigated the frequency of RFC1 intronic repeat expansions, a phenomenon implicated in CANVAS; a disease with a diagnostic overlap with MSA.[27][28] The study concluded that these repeats were absent in pathologically confirmed MSA, suggesting an alternative genetic cause.[27]
Pathophysiology
[edit]Multiple system atrophy can be explained as cell loss and gliosis or a proliferation of astrocytes in damaged areas of the central nervous system. This damage forms a scar which is then termed a glial scar.[29] The presence of inclusion bodies known as Papp–Lantos bodies, in the movement, balance, and autonomic-control centres of the brain are the defining histopathologic hallmark of MSA.[30]
The major filamentous component of Papp-Lantos bodies, glial and neuronal cytoplasmic inclusions, is alpha-synuclein.[31] Mutations in this substance may play a role in the disease.[32] The conformation of the alpha-synuclein is different from that of alpha-synuclein in Lewy bodies.[2] The disease probably starts with an oligodendrogliopathy.[33] It has been proposed that the α-synuclein inclusions found in Oligodendrocytes result from the pruning and the engulfment of diseased axonal segments containing aggregated α-synuclein, i.e., of Lewy neurites [34]
Tau proteins have been found in some glial cytoplasmic inclusion bodies.[35]
Diagnosis
[edit]Clinical
[edit]Clinical diagnostic criteria were defined in 1998[36] and updated in 2007[37] and in 2022.[38] Certain signs and symptoms of MSA also occur with other disorders, such as Parkinson's disease, making the diagnosis more difficult.[39][40][41]
Features characteristic of OPCA include progressive cerebellar ataxia, leading to clumsiness in body movements, veering from midline when walking, wide-based stance, and falls without signs of paralysis or weakness.[42][43] Clinical presentation can vary greatly between patients, but mostly affects speech, balance and walking.[44] Other possible neurological problems include spasmodic dysphonia, hypertonia, hyperreflexia, rigidity, dysarthria, dysphagia and neck dystonic posture.[43] Diagnosis may be based on a thorough medical exam; the presence of signs and symptoms; imaging studies; various laboratory tests; and an evaluation of the family history.[45]
Radiologic
[edit]Both MRI and CT scanning may show a decrease in the size of the cerebellum and pons in those with cerebellar features (MSA-C). The putamen is hypointense on T2-weighted MRI and may show an increased deposition of iron in the Parkinsonian (MSA-P) form. In MSA-C, a "hot cross bun" sign is sometimes found; it reflects atrophy of the pontocerebellar tracts that give T2 hyper intense signal intensity in the atrophic pons.
MRI changes are not required to diagnose the disease as these features are often absent, especially early in the course of the disease. Additionally, the changes can be quite subtle and are usually missed by examiners who are not experienced with MSA.[citation needed]
Pathologic
[edit]Pathological diagnosis can only be made at autopsy by finding abundant glial cytoplasmic inclusions (GCIs) on histological specimens of the central nervous system.[46]
Olivopontocerebellar atrophy can be used as a pathological term to describe degeneration of neurons in specific areas of the brain – the cerebellum, pons, and inferior olivary nucleus.[47] OPCA is present in several neurodegenerative syndromes, including inherited and non-inherited forms of ataxia (such as the hereditary spinocerebellar ataxia known as Machado–Joseph disease) and MSA, with which it is primarily associated.[47]
Contrary to most other synucleinopathies, which develop α-synuclein inclusions primarily in neuronal cell populations,[48] MSA presents with extensive pathological α-synuclein inclusions in the cytosol of oligodendrocytes (glial cytoplasmic inclusions), with limited pathology in neurons.[49] MSA also differs from other synucleinopathies in its regional pathological presentation, with α-synuclein positive inclusions detected predominantly in the striatum, midbrain, pons, medulla and cerebellum,[50][51] rather than the brainstem, limbic and cortical regions typically effected in Lewy inclusion diseases.[51] However, recent studies using novel, monoclonal antibodies specific for C-terminally truncated α-synuclein (αSynΔC) have now shown that neuronal α-synuclein pathology is more abundant than previously thought.[52][53] One group revealed robust α-synuclein pathology in the pontine nuclei and medullary inferior olivary nucleus upon histological analysis of neurological tissue from MSA patients.[52] Histopathological investigation on six cases of pathologically confirmed MSA, using antibodies directed at a variety of α-synuclein epitopes, revealed substantial variation in α-synuclein protein deposition across both cases and brain regions within cases, providing evidence for 'strains' of aggregated conformers that may differentially promote pathological prion-like spread.[54]
In 2020, researchers at The University of Texas Health Science Center at Houston concluded that protein misfolding cyclic amplification could be used to distinguish between two progressive neurodegenerative diseases, Parkinson's disease and multiple system atrophy, being the first process to give an objective diagnosis of Multiple System Atrophy instead of just a differential diagnosis.[55][56]
Classification
[edit]MSA is one of several neurodegenerative diseases known as synucleinopathies: they have in common an abnormal accumulation of alpha-synuclein protein in various parts of the brain. Other synucleinopathies include Parkinson's disease, the Lewy body dementias, and other more rare conditions.[57]
Old terminology
[edit]| Olivopontocerebellar atrophy | |
|---|---|
| Other names | Multiple system atrophy – cerebellar subtype[58] |
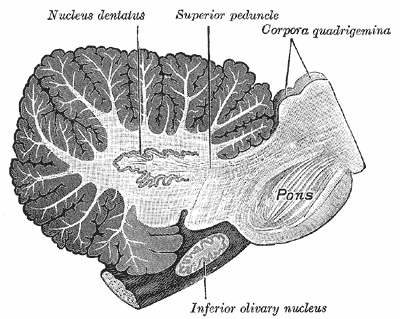 | |
| Sagittal section through right cerebellar hemisphere. The right olive has also been cut sagittally. | |
| Specialty | Neurology |
Historically, many terms were used to refer to this disorder, based on the predominant systems presented. These terms were discontinued by consensus in 1996 and replaced with MSA and its subtypes,[59] but awareness of these older terms and their definitions is helpful to understanding the relevant literature prior to 1996. These include striatonigral degeneration (SND), olivopontocerebellar atrophy (OPCA), and Shy–Drager syndrome.[60] A table describing the characteristics and modern names of these conditions follows:
| Historical Name | Characteristics | Modern name and abbreviation |
|---|---|---|
| Striatonigral degeneration | predominating Parkinson's-like symptoms | MSA-P, "p" = parkinsonian subtype |
| Sporadic olivopontocerebellar atrophy (OPCA) | characterized by progressive ataxia (an inability to coordinate voluntary muscular movements) of the gait and arms and dysarthria (difficulty in articulating words) | MSA-C, "c" = cerebellar dysfunction subtype |
| Shy-Drager syndrome | characterized by Parkinsonism plus a more pronounced failure of the autonomic nervous system.[61] | No modern equivalent – this terminology fell out of favour[62] and was not specified in the 2007 consensus paper.[37] The earlier consensus of 1998[36] referred to MSA-A, "a" = autonomic dysfunction subtype but this subtype is no longer used. |
The term olivopontocerebellar atrophy was originally coined by Joseph Jules Dejerine and André Thomas.[63][64] It was subdivided as:
| Number | OMIM | Alt. name | Inheritance |
|---|---|---|---|
| OPCA type 2 | 258300 | Fickler[65]-Winkler[66] type OPCA | autosomal recessive |
| OPCA type 5 | 164700 | OPCA with dementia and extrapyramidal signs | autosomal dominant |
Non-hereditary diseases formerly categorized as olivopontocerebellar atrophy have were reclassified as forms of MSA[67] as well as to four hereditary types, that have been currently reclassified as four different forms of spinocerebellar ataxia:
| Hereditary OPCA type | OPCA name | SCA # | Gene | OMIM |
|---|---|---|---|---|
| OPCA type 1 | "Menzel type OPCA" | SCA1 | ATXN1 | 164400 |
| OPCA type 2, autosomal dominant | "Holguin type OPCA" | SCA2 | ATXN2 | 183090 |
| OPCA type 3 | "OPCA with retinal degeneration" | SCA7 | ATXN7 | 164500 |
| OPCA type 4 | "Schut-Haymaker type OPCA" | SCA1 | ATXN1 | 164400 |
Current terminology
[edit]The current terminology and diagnostic criteria for the disease were established at a 2007 conference of experts and set forth in a position paper.[37] This Second Consensus Statement defines two categories of MSA, based on the predominant symptoms of the disease at the time of evaluation. These are:
- MSA with predominant parkinsonism (MSA-P) - defined as MSA where extrapyramidal features predominate. It is sometimes termed striatonigral degeneration, a parkinsonian variant.[citation needed]
- MSA with cerebellar features (MSA-C) - defined as MSA in which cerebellar ataxia predominates. It is sometimes termed sporadic olivopontocerebellar atrophy.[citation needed]
Management
[edit]Supervision
[edit]Ongoing care from a neurologist specializing in movement disorders is recommended,[by whom?] because the complex symptoms of MSA are often not familiar to less-specialized neurologists. Hospice/homecare services can be very useful as disability progresses.[citation needed]
Drug therapy
[edit]Levodopa (L-Dopa), a drug used in the treatment of Parkinson's disease, improves parkinsonian symptoms in a small percentage of MSA patients. A recent trial reported that only 1.5% of MSA patients experienced any improvement at all when taking levodopa, their improvement was less than 50%, and even that improvement was a transient effect lasting less than one year. Poor response to L-Dopa has been suggested as a possible element in the differential diagnosis of MSA from Parkinson's disease.[68]
The drug riluzole is ineffective in treating MSA or PSP.[9]
Rehabilitation
[edit]Management by rehabilitation professionals including physiatrists, physiotherapists, occupational therapists, speech therapists, and others for difficulties with walking/movement, daily tasks, and speech problems is essential.[citation needed]
Physiotherapists can help to maintain the patient's mobility and will help to prevent contractures.[29] Instructing patients in gait training will help to improve their mobility and decrease their risk of falls.[69] A physiotherapist may also prescribe mobility aids such as a cane or a walker to increase the patient's safety.[69]
Speech therapists may assist in assessing, treating and supporting speech (dysarthria) and swallowing difficulties (dysphagia). Speech changes mean that alternative communication may be needed, for example, communication aids or word charts.[citation needed]
Early intervention of swallowing difficulties is particularly useful to allow for discussion around tube feeding further in the disease progression.[citation needed] At some point in the progression of the disease, fluid and food modification may be implemented.[citation needed]
Avoidance of postural hypotension
[edit]One particularly serious problem, the drop in blood pressure upon standing up (with risk of fainting and thus injury from falling), often responds to fludrocortisone, a synthetic mineralocorticoid.[70][71] Another common drug treatment is the alpha-agonist midodrine.[70]
Non-drug treatments include "head-up tilt" (elevating the head of the whole bed by about 10 degrees), salt tablets or increasing salt in the diet, generous intake of fluids, and pressure (elastic) stockings. Avoidance of triggers of low blood pressure, such as hot weather, alcohol, and dehydration, are crucial.[71] The patient can be taught to move and transfer from sitting to standing slowly to decrease risk of falls and limit the effect of postural hypotension.[69] Instruction in ankle pumping helps to return blood in the legs to the systemic circulation.[69] Other preventative measures are raising the head of the bed by 8 in (20.3 cm), and the use of compression stockings and abdominal binders.[5]
Supine hypertension
[edit]In addition to orthostatic hypotension, supine hypertension, where the BP is excessively high lying down, is a frequent problem in multiple system atrophy. Treatment of one symptom can easily aggravate the other, and supine hypertension in such patients has been linked to the same cardiovascular complications as essential hypertension.[72]
Support
[edit]Social workers and occupational therapists can also help with coping with disability through the provision of equipment and home adaptations, services for caregivers and access to healthcare services, both for the person with MSA as well as family caregivers.[citation needed]
Prognosis
[edit]The average lifespan after the onset of symptoms in patients with MSA is 6–10 years.[3] Approximately 60% of patients require a wheelchair within five years of onset of the motor symptoms, and few patients survive beyond 12 years.[3] The disease progresses without remission at a variable rate. Those who present at an older age, those with parkinsonian features, and those with severe autonomic dysfunction have a poorer prognosis.[3] Those with predominantly cerebellar features and those who display autonomic dysfunction later have a better prognosis.[3]
Causes of death
[edit]The most common causes of death are sudden death and death caused by infections, which include urinary catheterization infections, feeding tube infections, and aspiration pneumonia. Some deaths are caused by cachexia, also known as wasting syndrome.[73]
Epidemiology
[edit]Multiple system atrophy is estimated to affect approximately 5 per 100,000 people. At autopsy, many patients diagnosed during life with Parkinson's disease are found actually to have MSA, suggesting that the actual incidence of MSA is higher than that estimate.[3] While some suggest that MSA affects slightly more men than women (1.3:1), others suggest that the two sexes are equally likely to be affected.[3][5][29] The condition most commonly presents in persons aged 50–60.[3]
Research
[edit]Mesenchymal stem cell therapy may delay the progression of neurological deficits in patients with MSA-cerebellar type.[74]
Notable cases
[edit]- Nikolai Andrianov was a Soviet/Russian gymnast who held the record for men for the most Olympic medals at 15 (7 gold medals, 5 silver medals, 3 bronze medals) until Michael Phelps surpassed him at the 2008 Beijing Summer Olympics.[75]
- Todd J. Campbell (1956–2021), United States district judge and counsel to former Vice President Al Gore.[76]
- Singer and songwriter Johnny Cash wrote in his autobiography that he was diagnosed with Shy–Drager in 1997.[77]
- Ronald Green (1944–2012), American-Israeli basketball player[78]
- Joseph C. Howard Sr. (1922-2000) was the first African American to serve as a United States district judge of the United States District Court for the District of Maryland.[79]
- Kenneth More British actor, originally diagnosed with Parkinson's disease.
- Chef Kerry Simon died from complications of MSA.[80]
- David Colin Sherrington FRS (1945–2014), noted polymer chemist, who was diagnosed in 2012 and died from pneumonia two years later.
- Karsten Heuer (1968-2024) Canadian Biologist, Conservationist, Filmmaker and Author.
See also
[edit]References
[edit]- ^ "Multiple system atrophy" at Dorland's Medical Dictionary
- ^ a b Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. (May 2018). "Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies". Nature. 557 (7706): 558–563. Bibcode:2018Natur.557..558P. doi:10.1038/s41586-018-0104-4. PMC 5970994. PMID 29743672.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b c d e f g h Fanciulli A, Wenning GK (January 2015). "Multiple-system atrophy". The New England Journal of Medicine. 372 (3): 249–263. doi:10.1056/NEJMra1311488. PMID 25587949.
- ^ "Multiple System Atrophy Clinical Presentation". Retrieved 7 January 2018.
- ^ a b c Swan L, Dupont J (May 1999). "Multiple system atrophy". Physical Therapy. 79 (5): 488–494. doi:10.1093/ptj/79.5.488. PMID 10331752.
- ^ Burn DJ, Jaros E (December 2001). "Multiple system atrophy: cellular and molecular pathology". Molecular Pathology. 54 (6): 419–426. PMC 1187133. PMID 11724918.
- ^ [unreliable medical source?]Sikorska B, Papierz W, Preusser M, Liberski PP, Budka H (February 2007). "Synucleinopathy with features of both multiple system atrophy and dementia with Lewy bodies". Neuropathology and Applied Neurobiology. 33 (1): 126–129. doi:10.1111/j.1365-2990.2006.00817.x. PMID 17239015. S2CID 40186391.
- ^ Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, et al. (July 2015). "Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with α-synuclein". Acta Neuropathologica. 130 (1): 93–105. doi:10.1007/s00401-015-1442-z. PMC 6764097. PMID 25962793.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN (January 2009). "Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study". Brain. 132 (Pt 1): 156–171. doi:10.1093/brain/awn291. PMC 2638696. PMID 19029129.
- ^ Oertel WH, Wächter T, Quinn NP, Ulm G, Brandstädter D (April 2003). "Reduced genital sensitivity in female patients with multiple system atrophy of parkinsonian type". Movement Disorders. 18 (4): 430–432. doi:10.1002/mds.10384. PMID 12671951. S2CID 28102026.
- ^ Stępnicki (20 August 2018). "Current Concepts and Treatments of Schizophrenia". Molecules. 23 (8): 2087. doi:10.3390/molecules23082087. ISSN 1420-3049. PMC 6222385. PMID 30127324.
- ^ Aminoff MJ, Greenberg DA, Simon RP (2005). "Chapter 7: Movement disorders". Clinical Neurology (6th ed.). Lange: McGraw-Hill Medical. pp. 241–45. ISBN 978-0-07-142360-1.
- ^ Ogawa T, Fujii S, Kuya K, Kitao SI, Shinohara Y, Ishibashi M, et al. (September 2018). "Role of Neuroimaging on Differentiation of Parkinson's Disease and Its Related Diseases". Yonago Acta Medica (Review). 61 (3): 145–155. doi:10.33160/yam.2018.09.001. PMC 6158357. PMID 30275744.
Parkinsonian syndromes are a group of movement disorders characterized by classical motor symptoms such as tremors, bradykinesia, and rigidity. They are most frequently due to primary neurodegenerative disease, resulting in the loss of dopaminergic nerve terminals along the nigrostriatal pathway, similar to idiopathic PD, MSA, PSP, CBD, and DLB.
- ^ Hodos W (2009). "Evolution of Cerebellum". Encyclopedia of Neuroscience. Springer. pp. 1240–1243. doi:10.1007/978-3-540-29678-2_3124. ISBN 978-3-540-23735-8.
- ^ "Autonomic nervous system" at Dorland's Medical Dictionary
- ^ "Hypotension". The Lecturio Medical Concept Library. Retrieved 27 July 2021.
- ^ Ackley B (2010). Nursing diagnosis handbook : an evidence-based guide to planning care (9th ed.). Maryland Heights, Mo: Mosby. ISBN 9780323071505.
- ^ Richard C, Amarenco G, Palma JA, Kaufmann H, Drapier S, Gamé X, et al. (2019). "Early bladder dysfunction in multiple system atrophy: who seek shall find". Clin Auton Res. 29 (6): 625–6. doi:10.1007/s10286-019-00648-2. PMID 1705345.
- ^ Sakakibara R, Panicker J, Finazzi-Agro E, Iacovelli V, Bruschini H, Subcomittee PD, et al. (2016). "A guideline for the management of bladder dysfunction in Parkinson's disease and other gait disorders". Neurourol Urodyn. 35 (5): 551–63. doi:10.1002/nau.22764. PMID 25810035.
- ^ Cunningham GR, Rosen RC (2018). "Overview of male sexual dysfunction.". In Martin KA (ed.). UpToDate. Waltham, MA: UpToDate.
- ^ "Constipation". The Lecturio Medical Concept Library. Retrieved 27 July 2021.
- ^ Gilman S, Koeppe RA, Chervin RD, Consens FB, Little R, An H, et al. (July 2003). "REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA". Neurology. 61 (1): 29–34. doi:10.1212/01.wnl.0000073745.68744.94. PMID 12847152. S2CID 9538306.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b "What is multiple system atrophy?". NIH. Retrieved 25 November 2018.
- ^ Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, et al. (August 2010). "Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy". Brain. 133 (Pt 8): 2382–2393. doi:10.1093/brain/awq158. PMID 20576697.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Sasaki H, Emi M, Iijima H, Ito N, Sato H, Yabe I, et al. (June 2011). "Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy". Molecular Brain. 4: 24. doi:10.1186/1756-6606-4-24. PMC 3141657. PMID 21658278.
Copy number loss of SHC2 strongly indicates a causal link to MSA.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Ferguson MC, Garland EM, Hedges L, Womack-Nunley B, Hamid R, Phillips JA, et al. (February 2014). "SHC2 gene copy number in multiple system atrophy (MSA)". Clinical Autonomic Research. 24 (1): 25–30. doi:10.1007/s10286-013-0216-8. PMC 3946192. PMID 24170347.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b Sullivan R, Yau WY, Chelban V, Rossi S, O'Connor E, Wood NW, et al. (July 2020). "RFC1 Intronic Repeat Expansions Absent in Pathologically Confirmed Multiple Systems Atrophy". Movement Disorders. 35 (7): 1277–1279. doi:10.1002/mds.28074. PMID 32333430. S2CID 216129457.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. (April 2019). "Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia". Nature Genetics. 51 (4): 649–658. doi:10.1038/s41588-019-0372-4. PMC 6709527. PMID 30926972.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b c Wenning GK, Colosimo C, Geser F, Poewe W (February 2004). "Multiple system atrophy". The Lancet. Neurology. 3 (2): 93–103. doi:10.1016/S1474-4422(03)00662-8. PMID 14747001. S2CID 10162139.
Wenning GK, Colosimo C, Geser F, Poewe W (March 2004). "Erratum". Lancet Neurol. 3 (3): 137. doi:10.1016/S1474-4422(04)00695-7. S2CID 208782339. - ^ Jellinger KA, Lantos PL (June 2010). "Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update". Acta Neuropathologica. 119 (6): 657–667. doi:10.1007/s00401-010-0672-3. PMID 20309568. S2CID 19759468.
- ^ Arima K, Uéda K, Sunohara N, Arakawa K, Hirai S, Nakamura M, et al. (November 1998). "NACP/alpha-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy". Acta Neuropathologica. 96 (5): 439–444. doi:10.1007/s004010050917. PMID 9829806. S2CID 10804119.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Al-Chalabi A, Dürr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, et al. (September 2009). "Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy". PLOS ONE. 4 (9): e7114. Bibcode:2009PLoSO...4.7114A. doi:10.1371/journal.pone.0007114. PMC 2743996. PMID 19771175.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Stefanova N, Wenning GK (February 2016). "Review: Multiple system atrophy: emerging targets for interventional therapies". Neuropathology and Applied Neurobiology. 42 (1): 20–32. doi:10.1111/nan.12304. PMC 4788141. PMID 26785838.
- ^ De Nuccio F, Kashyrina M, Serinelli F, Laferrière F, Lofrumento DD, De Giorgi F, et al. (February 2023). "Oligodendrocytes Prune Axons Containing α-Synuclein Aggregates In Vivo: Lewy Neurites as Precursors of Glial Cytoplasmic Inclusions in Multiple System Atrophy?". Biomolecules. 13 (2): 269. doi:10.3390/biom13020269. PMC 9953613. PMID 36830639.
- ^ Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, et al. (March 2001). "Co-localization of alpha-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration". Acta Neuropathologica. 101 (3): 285–293. doi:10.1007/s004010000292. PMID 11307630. S2CID 25650403.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, et al. (February 1999). "Consensus statement on the diagnosis of multiple system atrophy". Journal of the Neurological Sciences. 163 (1): 94–98. doi:10.1016/s0022-510x(98)00304-9. hdl:2027.42/41757. PMID 10223419. S2CID 13307970.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b c Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. (August 2008). "Second consensus statement on the diagnosis of multiple system atrophy". Neurology. 71 (9): 670–676. doi:10.1212/01.wnl.0000324625.00404.15. PMC 2676993. PMID 18725592.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, et al. (2022). "The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy". Movement Disorders. 37 (6): 1131–48. doi:10.1002/mds.29005. hdl:11585/899814. PMC 9321158. PMID 35445419.
- ^ "Multiple System Atrophy / Shy Drager Syndrome". Vanderbilt Autonomic Dysfunction Center. Retrieved 29 May 2010.
- ^ Bloomfield SM, Hanna PA, Noor ER, Dalvi AI (24 September 2018). Benbadis SR (ed.). "multiple system atrophy overview". Medscape. WebMD LLC.
- ^ Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, et al. (August 2015). "When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients". Neurology. 85 (5): 404–412. doi:10.1212/WNL.0000000000001807. PMC 4534078. PMID 26138942.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Landers M, Adams M, Acosta K, Fox A. (2009). "Challenge-oriented gait and balance training in sporadic olivopontocerebellar atrophy: a case study". J Neurol Phys Ther. 33 (3): 160–168. doi:10.1097/npt.0b013e3181b511f4. PMID 19809395. S2CID 24642594.
- ^ a b Berciano J, Boesch S, Pérez-Ramos JM, Wenning GK (2006). "Olivopontocerebellar atrophy: toward a better nosological definition". Mov. Disord. 21 (10): 1607–13. doi:10.1002/mds.21052. PMID 16874757. S2CID 38376147.
- ^ "Olivopontocerebellar atrophy - PubMed Health". Archived from the original on 17 November 2012. Retrieved 7 September 2017.
- ^ "Olivopontocerebellar atrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program".
- ^ Papp MI, Kahn JE, Lantos PL (December 1989). "Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome)". Journal of the Neurological Sciences. 94 (1–3): 79–100. doi:10.1016/0022-510X(89)90219-0. PMID 2559165. S2CID 1199951.
- ^ a b "NINDS Olivopontocerebellar Atrophy Information Page". Archived from the original on 27 January 2012. Retrieved 7 February 2012.
- ^ Waxman EA, Giasson BI (July 2009). "Molecular mechanisms of alpha-synuclein neurodegeneration". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1792 (7): 616–624. doi:10.1016/j.bbadis.2008.09.013. PMC 2756732. PMID 18955133.
- ^ Burn DJ, Jaros E (December 2001). "Multiple system atrophy: cellular and molecular pathology". Molecular Pathology. 54 (6): 419–426. PMC 1187133. PMID 11724918.
- ^ Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. (December 2004). "The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations". Brain. 127 (Pt 12): 2657–2671. doi:10.1093/brain/awh303. PMID 15509623.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b Brettschneider J, Suh E, Robinson JL, Fang L, Lee EB, Irwin DJ, et al. (November 2018). "Converging Patterns of α-Synuclein Pathology in Multiple System Atrophy". Journal of Neuropathology and Experimental Neurology. 77 (11): 1005–1016. doi:10.1093/jnen/nly080. PMC 6181179. PMID 30203094.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b Hass EW, Sorrentino ZA, Lloyd GM, McFarland NR, Prokop S, Giasson BI (May 2021). "Robust α-synuclein pathology in select brainstem neuronal populations is a potential instigator of multiple system atrophy". Acta Neuropathologica Communications. 9 (1): 80. doi:10.1186/s40478-021-01173-y. PMC 8091528. PMID 33941284.
- ^ Hass EW, Sorrentino ZA, Xia Y, Lloyd GM, Trojanowski JQ, Prokop S, et al. (August 2021). "Disease-, region- and cell type specific diversity of α-synuclein carboxy terminal truncations in synucleinopathies". Acta Neuropathologica Communications. 9 (1): 146. doi:10.1186/s40478-021-01242-2. PMC 8403399. PMID 34454615.
- ^ Dhillon JS, Trejo-Lopez JA, Riffe C, McFarland NR, Hiser WM, Giasson BI, et al. (July 2019). "Dissecting α-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis". Laboratory Investigation; A Journal of Technical Methods and Pathology. 99 (7): 982–992. doi:10.1038/s41374-019-0198-9. PMC 7209695. PMID 30737468.
- ^ "Method Can Distinguish Parkinson's Disease From multiple system atrophy". Diagnostics from Technology Networks. Retrieved 23 February 2020.
- ^ Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. (February 2020). "Discriminating α-synuclein strains in Parkinson's disease and multiple system atrophy". Nature. 578 (7794): 273–277. Bibcode:2020Natur.578..273S. doi:10.1038/s41586-020-1984-7. PMC 7066875. PMID 32025029.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Goedert M, Jakes R, Spillantini MG (2017). "The Synucleinopathies: Twenty Years On". Journal of Parkinson's Disease (Review). 7 (s1): S51–S69. doi:10.3233/JPD-179005. PMC 5345650. PMID 28282814.
- ^ "Multiple system atrophy – cerebellar subtype: MedlinePlus Medical Encyclopedia". medlineplus.gov. Retrieved 24 May 2019.
- ^ The Consensus Committee of the American Autonomic Society and the American Academy of Neurology (May 1996). "Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy". Neurology. 46 (5): 1470. doi:10.1212/wnl.46.5.1470. PMID 8628505. S2CID 219212717.
- ^ Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, et al. (February 2012). "The neuropathology, pathophysiology and genetics of multiple system atrophy". Neuropathology and Applied Neurobiology. 38 (1): 4–24. doi:10.1111/j.1365-2990.2011.01234.x. PMID 22074330. S2CID 22901422.
- ^ Shy GM, Drager GA (May 1960). "A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study". Archives of Neurology. 2 (5): 511–527. doi:10.1001/archneur.1960.03840110025004. PMID 14446364.
- ^ Schatz IJ (July 1996). "Farewell to the "Shy-Drager syndrome"". Annals of Internal Medicine. 125 (1): 74–75. doi:10.7326/0003-4819-125-1-199607010-00012. PMID 8644992. S2CID 8594266.
- ^ synd/1903 at Who Named It? - "Dejerine-Thomas atrophy"
- ^ J. J. Dejerine, A. Thomas. L’atrophie olivo-ponto-cérébelleuse. Nouvelle iconographie de la Salpêtrière, Paris, 1900, 13: 330-370. 1912, 25: 223-250.
- ^ Fickler, A. Klinische und pathologisch-anatomische Beitraege zu den Erkrankungen des Kleinhirns. Dtsch. Z. Nervenheilk. 41: 306-375, 1911.
- ^ Winkler, C. A case of olivo-pontine cerebellar atrophy and our conceptions of neo- and palaio-cerebellum. Schweiz. Arch. Neurol. Psychiat. 13: 684-702, 1923.
- ^ MeSH Result
- ^ Calandra-Buonaura G, Doria A, Lopane G, Guaraldi P, Capellari S, Martinelli P, et al. (February 2016). "Pharmacodynamics of a low subacute levodopa dose helps distinguish between multiple system atrophy with predominant Parkinsonism and Parkinson's disease". Journal of Neurology. 263 (2): 250–256. doi:10.1007/s00415-015-7961-7. PMID 26566913. S2CID 189866517.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ a b c d Hardy J (2008). "Multiple system atrophy: pathophysiology, treatment and nursing care". Nursing Standard. 22 (22): 50–6, quiz 58. doi:10.7748/ns2008.02.22.22.50.c6359. PMID 18333558.
- ^ a b Multiple system atrophy (MSA) mayoclinic.org, accessed 20 May 2018
- ^ a b Palma JA, Kaufmann H (February 2020). "Management of Orthostatic Hypotension". Continuum. 26 (1): 154–177. doi:10.1212/CON.0000000000000816. PMC 7339914. PMID 31996627.
- ^ Palma JA, Redel-Traub G, Porciuncula A, Samaniego-Toro D, Millar Vernetti P, Lui YW, et al. (June 2020). "The impact of supine hypertension on target organ damage and survival in patients with synucleinopathies and neurogenic orthostatic hypotension". Parkinsonism & Related Disorders. 75 (75): 97–104. doi:10.1016/j.parkreldis.2020.04.011. PMC 7415666. PMID 32516630.
- ^ Papapetropoulos S, Tuchman A, Laufer D, Papatsoris AG, Papapetropoulos N, Mash DC (March 2007). "Causes of death in multiple system atrophy". Journal of Neurology, Neurosurgery, and Psychiatry. 78 (3): 327–329. doi:10.1136/jnnp.2006.103929. PMC 2117630. PMID 17308296.
- ^ Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, et al. (July 2012). "A randomized trial of mesenchymal stem cells in multiple system atrophy". Annals of Neurology. 72 (1): 32–40. doi:10.1002/ana.23612. PMID 22829267. S2CID 5201446.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Turner A. "Olympic Legend Andrianov Dies at 58". International Gymnast Magazine Online. Retrieved 10 December 2018.
- ^ Hubbard A, Timms M. "Former U.S. District Judge Todd Campbell, longtime Nashville legal mind and adviser to a vice president, dead at 64". The Tennessean.
- ^ Cash J, Carr P (1998) [1997]. Cash: The Autobiography. New York, NY, USA: HarperCollins Publishers. pp. 400–403. ISBN 978-0061013577.
- ^ "Ronald Green Obituary". The Miami Herald. 26 July 2012.
- ^ Cummings E (30 September 2000). "Standing up for justice". Baltimore AFRO-American. Archived from the original on 4 May 2008. Retrieved 10 December 2018.
- ^ "Kerry Simon, Las Vegas 'Iron Chef' winner, dies at 60". Business Insider. Archived from the original on 14 November 2018. Retrieved 14 November 2018.
External links
[edit]- Medical Textbook: "Multiple System Atrophy" edited by Gregor Wenning and Alessandra Fanciulli