Salt and pepper syndrome
| Salt and pepper syndrome | |
|---|---|
| Other names | Amish infantile epilepsy syndrome, ST3GAL5-CDG, Salt and pepper mental retardation syndrome, Infantile-onset symptomatic epilepsy syndrome - developmental stagnation - blindness, Epilepsy syndrome, infantile-onset symptomatic[1][2][3] |
 | |
| Specialty | Medical genetics |
| Usual onset | Childhood |
| Causes | Genetic mutation |
| Prevention | none |
Salt and pepper developmental regression syndrome, also known as Amish infantile epileptic syndrome[4] or GM3 deficiency syndrome,[5] is a rare autosomal recessive progressive neurological disorder characterized by developmental delay, severe intellectual disability, seizures, and skin pigmentation irregularities.[6][7][8][9] The clinical symptoms of this condition start manifesting soon after birth, during the newborn/neo-natal stage of life.[10]
Signs and symptoms
[edit]The clinical course of this condition, as mentioned before, typically starts soon after birth.[citation needed]
The first features of the syndrome start off with feeding difficulties, constant irritability, frequent vomiting, and hypotonia.[11][6][10]
The second manifestations of this condition are frequent seizures whose type can vary between patients, but usually consist of tonic-clonic seizures. These usually start sometime during the baby's first year of life.[11][6][10]
As the disease progresses, intellectual abilities, developmental milestones, and the senses of hearing and vision start deteriorating, this pathology will cause a phenomenon known as developmental regression (a condition where all developmental abilities a baby has acquired through their short life-time start deteriorating), severe intellectual disabilities, and will eventually lead to severe visual and hearing impairments in some to most patients.[11][6][10]
Some patients with the condition start developing involuntary movements of the arms and/or legs that are similar to those shown by patients with choreoathetosis.[11][6][10]
Another relatively benign symptom shown by patients with this condition is localized hyperpigmentation spots, which resemble freckles and are usually spread all over a patient's body, there is no accurate way of predicting whether these spots will fade or not in a patient. As previously mentioned, these cutaneous spots are benign, meaning that they don't cause any symptoms in and on themselves.[11][6][10]
Rare features of the disorder include self-harming behaviors and icthyosis.[8]
Complications
[edit]The feeding difficulties and regular vomiting characteristic of this condition cause failure to thrive,[12] a condition characterized by lower-than-average weight gain in babies and toddlers which can in and on itself cause even more complications, such as malnutrition.[medical citation needed]
Genetics
[edit]This condition is caused by a mutation known under the name of Arg288Ter (also known as R288X), said mutation is found in the ST3GAL5 gene, located on the chromosome 2. ST3GAL5 is responsible for the production of an enzyme known as GM3 synthase, this enzyme is essential for the creation of a group of molecules known as gangliosides,[13] which, although present in almost all tissues and cells of the body, are mostly concentrated on the nervous system.[14] The mutation involved in this condition results in premature termination of GM3 synthase which essentially results in a disruption affecting the production of GM3 synthase, the absence of this enzyme significantly interrupts the production of any ganglioside,[11][15] which in turn doesn't let the brain develop and function properly.[16] The reason as to why the absence of gangliosides in children with the disorder causes abnormal brain development is still unknown.[14]
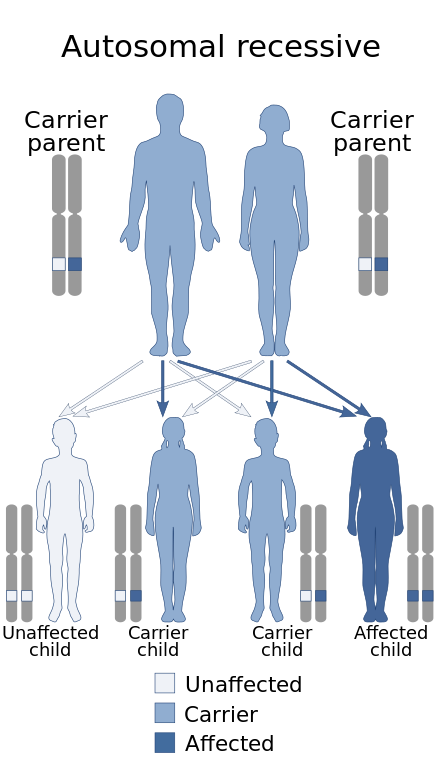
These mutations are inherited in an autosomal recessive fashion, which means that for a child to show the signs of the disorder, both of their parents must have transmitted them at least one copy of the dysfunctional ST3GAL5 gene. This transmission can occur whether or not the parents are affected with the disease themselves or not. If the parents are heterozygous (meaning they only carry one copy of the mutation) carriers of the gene for the disorder, there is a 1 in 4 (or 25%) chance of one of their offspring inheriting both copies of the defective gene which would make the child a homozygous carrier of the mutation, consequently giving the child the disorder.[17][18]
Diagnosis
[edit]This condition can be diagnosed through genetic testing methods such as whole exome sequencing or targeted sequencing of the ST3GAL5 gene for mutations, MRIs, electroencephalograms, IQ tests, and physical examination.[citation needed]
Genetic testing
[edit]Genetic testing is a very essential part of diagnosing salt pepper syndrome, without a genetic test, the only proof one would have for a diagnosis of the syndrome would be physical and intellectual anomalies, which wouldn't be sufficient to warrant a diagnosis.[citation needed]
Jamal et al. (2021) found a homozygous frameshift mutation (c.1030_1031del) in the ST3GAL5 gene of an Iranian child with the disorder born to consanguineous, first-cousin parents.[19]
Luigi et al. (2014) found a homozygous missense mutation (c.994G>A, also called p.E332K) in the ST3GAL5 gene of two American siblings previously described in the medical literature through the genetic testing methods of whole exome sequencing and Sanger sequencing.[20]
Jin et al. (2016) found compound heterozygous missense mutations through whole exome sequencing and Sanger sequencing in two South Korean sisters who were originally thought to have a Rett-like syndrome.[21]
MRIs
[edit]In 2013, Konstantina et al. described 2 siblings, born of consanguineous French parents. The both of them showed signs of salt pepper syndrome. Brain MRIs done on them came back with results showing cortical atrophy and lesions of the brain's white matter.[22]
Electroencephalograms
[edit]Electroencephalograms done on the affected patients reported by Simpson et al. showed "multifocal epileptiform discharges" which were "superimposed on a diffuse slow background activity."[23]
IQ tests
[edit]An IQ score of less than 70 obtained through an IQ test is typically sufficient to warrant a diagnosis of intellectual disability in children with the condition.[24]
Physical examination
[edit]Physical examination done in a clinical environment is also necessary for a diagnosis of salt & pepper syndrome.[citation needed]
Ophthalmoscopy
[edit]In 2006, Fahhad et al. published the results of an ophthalmoscopy study done on 4 children from 2 sibships of an Amish family; they showed optic nerve paleness on both eyes, indicative of bilateral optic atrophy, a condition which can lead to visual impairment and is usually associated with other disorders (including salt pepper syndrome).[25]
Anomaly scan
[edit]Anomaly scan is a prenatal diagnostic method which can be used to detect congenital anomalies in fetuses under ultrasound.[citation needed]
Jamal et al. (2021) found microcephaly on a child with the disorder prior to her birth using this diagnostic method.[19]
Treatment
[edit]Feeding difficulties
[edit]The parents of the patients with SPS reported by Simpson et al. used gastric tube feeding to feed their affected children, this treatment method is usually needed by most children with the disorder due to their feeding difficulties.[23]
Seizures
[edit]Seizures shown by patients with this condition cannot be treated with conventional anticonvulsant medication.[citation needed]
A study done in 2019 by Bowser et al. on 50 Amish patients with the condition showed that prescribed antiseizure medications such as phenobarbital, benzodiazepines, and valproic acid had some effect at treating the patients' seizures, insomnia, and irritability, which helped decrease their parents' overall stress of taking care of them.[26]
2 of the children reported by Simpson et al. had their seizures managed with vagus nerve stimulators.[23]
Prevalence
[edit]According to OMIM, around 100-200 cases of salt and pepper syndrome have been described in the medical literature. Most of these cases were from very large Old Order Amish families in Pennsylvania and Ohio.[27] The mutations are an example of the Founder effect in action.[28]
As of 2022, only 26 cases from non-Amish patients affected with salt & pepper syndrome have been described in the medical literature.[28]
History
[edit]This condition was first discovered in 2004 by Simpson et al. Their patients were 9 members from 2 sibships belonging to a single large, endogamous Amish family from Geauga County, Ohio. Their symptoms started between the ages of 2 weeks old and 3 months old, with irritability, vomiting, feeding difficulties and a failure to thrive followed by seizures in the first year of their life. They had developmental regression and couldn't do almost anything without the help of someone else. They had choreoathetosic non-purposeful hand movements. Brain studies on the oldest of the 9 affected members showed brain atrophy (which younger patients didn't have). Electroencephalograms gave back abnormal results. All members of the family shared a common great-great-grandparent. The disorder shown by this family clearly followed an autosomal recessive inheritance pattern. A homozygosity mapping study done afterwards on the affected family members showed linkage to the 2p12-p11.2 locus on chromosome 2, and after sequencing genes in said chromosomal region, they found a mutation of the nonsense type in their ST3GAL5 gene.[23]
A follow-up study done on the family in 2004 by Simpson, Cross, Proukakis, et.al., revealed that one of the studied members had died (nearly 3 years old) while the younger members had made it to their teens, with the oldest of them being 18 years old.[23]
Although salt and pepper developmental regression syndrome is prevalent among the Old Order Amish, further studies in 2016, 2021, and in 2023, found it in two Korean sisters, in an Iranian patient, and in a family in Saudi Arabia.[29][30][31]
Eponym
[edit]This condition has various names, but the three main ones are:[citation needed]
- Salt and pepper syndrome: Owing to the resemblance that a patient's cutaneous hypo/hyperpigmentation spots have to the condiment of either salt or pepper. This is the most commonly used name for the condition online.
- Amish infantile epileptic syndrome: Referencing both the concept of how common this condition is among Amish people (especially those who inhabit Pennsylvania and Ohio) and the infancy-onset epileptic seizures (alternative name for recurrent seizures) characteristic of this condition.
- GM3 deficiency syndrome: Name is pretty self-explanatory; it owes its name to the deficiency of GM3 synthase that patients with this condition suffer from.
References
[edit]- ^ "GM3 synthase deficiency". NORD (National Organization for Rare Disorders). Retrieved 2022-10-09.
- ^ "salt and pepper mental retardation syndrome | Hereditary Ocular Diseases". disorders.eyes.arizona.edu. Retrieved 2022-10-09.
- ^ "GM3 Synthase Deficiency via the ST3GAL5 Gene Test - PreventionGenetics". www.preventiongenetics.com. Retrieved 2022-10-10.
- ^ "Amish infantile epilepsy syndrome | Hereditary Ocular Diseases". disorders.eyes.arizona.edu. Retrieved 2022-10-09.
- ^ "GM3 Synthase Deficiency | Hereditary Ocular Diseases". disorders.eyes.arizona.edu. Retrieved 2022-10-09.
- ^ a b c d e f "GM3 synthase deficiency - About the Disease". Genetic and Rare Diseases Information Center. Retrieved 2022-10-09.
- ^ "UniProt". www.uniprot.org. Retrieved 2022-10-10.
- ^ a b Gordon-Lipkin E, Cohen JS, Srivastava S, Soares BP, Levey E, Fatemi A (November 2018). "ST3GAL5-Related Disorders: A Deficiency in Ganglioside Metabolism and a Genetic Cause of Intellectual Disability and Choreoathetosis". Journal of Child Neurology. 33 (13): 825–831. doi:10.1177/0883073818791099. PMC 6188822. PMID 30185102.
- ^ admin (2021-09-10). "Salt and Pepper Developmental Regression Syndrome (ST3GAL5)". Sema4. Retrieved 2022-10-10.
- ^ a b c d e f "GM3 synthase deficiency". Orphanet. Retrieved 2022-10-09.
- ^ a b c d e f "GM3 synthase deficiency". MedlinePlus Genetics. U.S. National Library of Medicine. Retrieved 2022-10-09.
- ^ "GM3 synthase deficiency". MedGen. NCBI, U.S. National Library of Medicine. C1836824. Retrieved 2022-10-10.
- ^ "GM3 Synthase Deficiency". UMass Chan Medical School. 2022-02-09. Retrieved 2022-10-09.
- ^ a b "ST3GAL5 gene". MedlinePlus Genetics. U.S. National Library of Medicine. Retrieved 2022-10-09.
- ^ "Kegg Disease: Amish infantile epilepsy syndrome". www.genome.jp. Retrieved 2022-10-10.
- ^ "GM3 Synthase Deficiency | Children's Hospital Pittsburgh". Children's Hospital of Pittsburgh. Retrieved 2022-10-09.
- ^ "Autosomal recessive inheritance pattern". Mayo Clinic. Retrieved 2022-10-09.
- ^ Cremaschi A (2021-08-03). "Autosomal recessive inheritance". Breda Genetics srl (in European Spanish). Retrieved 2022-10-09.
- ^ a b Manoochehri J, Dastgheib SA, Khamirani HJ, Mollaie M, Sharifi Z, Zoghi S, et al. (August 2021). "A novel frameshift pathogenic variant in ST3GAL5 causing salt and pepper developmental regression syndrome (SPDRS): A case report". Human Genome Variation. 8 (1): 33. doi:10.1038/s41439-021-00164-8. PMC 8361121. PMID 34385424.
- ^ Boccuto L, Aoki K, Flanagan-Steet H, Chen CF, Fan X, Bartel F, et al. (January 2014). "A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation". Human Molecular Genetics. 23 (2): 418–433. doi:10.1093/hmg/ddt434. PMC 3869362. PMID 24026681.
- ^ Lee JS, Yoo Y, Lim BC, Kim KJ, Song J, Choi M, Chae JH (August 2016). "GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype". American Journal of Medical Genetics. Part A. 170 (8): 2200–2205. doi:10.1002/ajmg.a.37773. PMID 27232954. S2CID 28838565.
- ^ Fragaki K, Ait-El-Mkadem S, Chaussenot A, Gire C, Mengual R, Bonesso L, et al. (May 2013). "Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency". European Journal of Human Genetics. 21 (5): 528–534. doi:10.1038/ejhg.2012.202. PMC 3641379. PMID 22990144.
- ^ a b c d e Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, et al. (November 2004). "Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase". Nature Genetics. 36 (11): 1225–1229. doi:10.1038/ng1460. PMID 15502825. S2CID 19516215.
- ^ "How do healthcare providers diagnose intellectual and developmental disabilities (IDDs)?". www.nichd.nih.gov/. Retrieved 2022-10-09.
- ^ Farukhi F, Dakkouri C, Wang H, Wiztnitzer M, Traboulsi EI (September 2006). "Etiology of vision loss in ganglioside GM3 synthase deficiency". Ophthalmic Genetics. 27 (3): 89–91. doi:10.1080/13816810600862626. PMID 17050284. S2CID 39175121.
- ^ Bowser LE, Young M, Wenger OK, Ammous Z, Brigatti KW, Carson VJ, et al. (April 2019). "Recessive GM3 synthase deficiency: Natural history, biochemistry, and therapeutic frontier". Molecular Genetics and Metabolism. 126 (4): 475–488. doi:10.1016/j.ymgme.2019.01.013. PMID 30691927. S2CID 59340942.
- ^ "Entry - #609056 - Salt and Pepper Developmental Regression Syndrome; SPDRS - OMIM". www.omim.org. Retrieved 2022-10-09.
- ^ a b Heide S, Jacquemont ML, Cheillan D, Renouil M, Tallot M, Schwartz CE, et al. (February 2022). "GM3 synthase deficiency in non-Amish patients". Genetics in Medicine. 24 (2): 492–498. doi:10.1016/j.gim.2021.10.007. PMID 34906476. S2CID 244793014.
- ^ Lee JS, Yoo Y, Lim BC, Kim KJ, Song J, Choi M, Chae JH (August 2016). "GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype". American Journal of Medical Genetics. Part A. 170 (8): 2200–2205. doi:10.1002/ajmg.a.37773. PMID 27232954. S2CID 28838565.
- ^ Borchers A, Pieler T (November 2010). "Programming pluripotent precursor cells derived from Xenopus embryos to generate specific tissues and organs". Genes. 1 (3). Creative Commons (CC BY): 413–426. doi:10.3390/genes14020354. PMC 3966229. PMID 24710095.
- ^ Manoochehri J, Dastgheib SA, Khamirani HJ, Mollaie M, Sharifi Z, Zoghi S, et al. (November 2021). "Correction: A novel frameshift pathogenic variant in ST3GAL5 causing salt and pepper developmental regression syndrome (SPDRS): a case report". Human Genome Variation. 8 (1): 42. doi:10.1038/s41439-021-00174-6. PMC 8608809. PMID 34811348.