Organorhodium chemistry

Organorhodium chemistry is the chemistry of organometallic compounds containing a rhodium-carbon chemical bond, and the study of rhodium and rhodium compounds as catalysts in organic reactions.[1]
Stable organorhodium compounds and transient organorhodium intermediates are used as catalyst such as in olefin hydroformylation, olefin hydrogenation, olefin isomerization and the Monsanto process.[2]
Classification based on principal oxidation states
[edit]Organometallic rhodium compounds share many characteristics with those of iridium, but less so with cobalt. Rhodium can exist in oxidation states of -III to +V, but rhodium(I) and rhodium(III) are the more common. Rhodium(I) compounds (d8 configuration) usually occur with square planar or trigonal bipyramidal geometries, while rhodium (III) compounds (d6 configuration) typically have an octahedral geometry.[2]
Rhodium(0)
[edit]Rhodium(0) complexes are binary carbonyls, the principal examples being tetrarhodium dodecacarbonyl, Rh4(CO)12, and hexadecacarbonylhexarhodium, Rh6(CO)16. These compounds are obtained by reductive carbonylation of rhodium(III) salts or Rh2Cl2(CO)4. In contrast to the stability of the homologous Co2(CO)8, Rh2(CO)8 is very labile.
Rhodium(I)
[edit]Rhodium(I) complexes are important homogeneous catalysts. Common complexes include bis(triphenylphosphine)rhodium carbonyl chloride, chlorobis(ethylene)rhodium dimer, cyclooctadiene rhodium chloride dimer, chlorobis(cyclooctene)rhodium dimer, dicarbonyl(acetylacetonato)rhodium(I), and rhodium carbonyl chloride. Although not formally organometallic, Wilkinson's catalyst (RhCl(PPh3)3), is included in the list of important catalysts. The simple olefin complexes chlorobis(ethylene)rhodium dimer, chlorobis(cyclooctene)rhodium dimer, and cyclooctadiene rhodium chloride dimer are often used as sources of "RhCl", exploiting the lability of the alkene ligands or their susceptibility to removal by hydrogenation. (η5-Cp)RhL2 are derived from Rh2Cl2L4 (L = CO, C2H4).
Rhodium(II)
[edit]Unlike the prevalence of cobalt(II) complexes, compounds of rhodium(II) are rare. The sandwich compound rhodocene is one example, even it exists in equilibrium with a dimeric Rh(I) derivative. Although not organometallic, rhodium(II) acetate (Rh2(OAc)4) catalyzes cyclopropanations via organometallic intermediates. Rhodium(II) porphyrin complexes react with methane.[3]
Rhodium(III)
[edit]Rhodium is usually supplied commercially in the Rh(III) oxidation state, the main starting reagent being hydrated rhodium trichloride. The latter reacts with olefins and with CO to give organometallic complexes, often concomitant with reduction to Rh(I). Cyclopentadienyl complexes of rhodium include the half-sandwich compound pentamethylcyclopentadienyl rhodium dichloride dimer.
Rhodium(V)
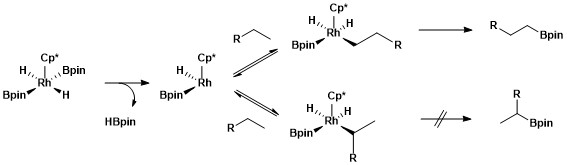
[edit]Strong donor ligands - hydride, silyl, boryl - are required to stabilize Rh(V). This oxidation state is invoked in borylation reactions.
Metallacycles
[edit]Cyclometalated rhodium compounds constitute an important class of organometallic chemistry. Although such compounds are well documented in the literature rhodium(III) cyclometalates with azo function are spare. A typical example of this category viz. novel hexacoordinated orthometalated rhodium(III) thiolato complex trans-[Rh(C∧N∧S)Cl(PPh3)2] was synthesized from benzyl 2-(phenylazo)phenyl thioether and RhCl3·3H2O in the presence of excess PPh3 via in situ C(sp2)−H and C(sp3)−S bond scissions. This is the first example for a coordination compound of (phenylazo)thiolate ligand. The mechanism of formation of orthometalated azobenzene derivative was described to proceed via initial coordination of azo-nitrogen followed by electrophilic substitution at the pendant phenyl ring. PPh3 plays a crucial role in the C(sp3)−S cleavage process. Reductive cleavage by single electron transfer (SET) mechanism is likely to be operative for the C−S bond cleavage. Unlike analogous (phenylazo)phenolato compound the orthometalated thiolato complex exhibits a fully reversible oxidative wave at 0.82 V vs Ag/AgCl and this response is supposed to be primarily centered on the thiolato sulfur atom.[4]
Main applications
[edit]Despite its high cost, rhodium is heavily relied on as a commercial catalyst.
Acetic acid and acetic anhydride syntheses
[edit]The Monsanto process is an industrial method for the making of acetic acid by catalytic carbonylation of methanol,[5] although it has largely been supplanted by the iridium-based Cativa process.

The catalytically active species is the anion cis-[Rh(CO)2I2]−.[6] which undergoes oxidative addition with methyl iodide. The related Tennessee Eastman acetic anhydride process affords acetic anhydride by carbonylation of methyl acetate.[7]
- CH3CO2CH3 + CO → (CH3CO)2O
Hydroformylation
[edit]
Hydroformylations often rely on rhodium-based catalysts. Water-soluble catalysts have also been developed. They facilitate the separation of the products from the catalyst.[8]
Hydrogenation
[edit]Wilkinson's catalyst is used as a homogeneous catalyst for the hydrogenation of olefins.[9] The mechanism of catalysis involves oxidative addition of H2, π-complexation of alkene, migratory insertion (intramolecular hydride transfer or olefin insertion), and reductive elimination.
Cationic organorhodium(I) catalysts are useful for asymmetric hydrogenations, which are applied to bioactive products such as pharmaceutical agents and agrochemicals.[10]

Other reactions
[edit]Nitrobenzene reduction is another reaction catalysed by this compound type:
References
[edit]- ^ Synthesis of Organometallic Compounds: A Practical Guide Sanshiro Komiya Ed. S. Komiya, M. Hurano 1997
- ^ a b Crabtree, Robert H. (2005). The Organometallic Chemistry of the Transition Metals (4th ed.). USA: Wiley-Interscience. ISBN 0-471-66256-9.
- ^ Bradford B. Wayland; Sujuan Ba; Alan E. Sherry (1991). "Activation of Methane and Toluene by Rhodium(II) Porphyrin Complexes". J. Am. Chem. Soc. 113 (14): 5305–5311. doi:10.1021/ja00014a025.
- ^ K. Pramanik; U. Das; B. Adhikari; D. Chopra; H. Stoeckli-Evans (2008). "RhCl3-Assisted C-H and C-S Bond Scissions: Isomeric Self-Association of Organorhodium(III) Thiolato Complex. Synthesis, Structure, and Electrochemistry". Inorg. Chem. 47 (2): 429–438. doi:10.1021/ic7016006. PMID 18161963.
- ^ Hosea Cheung, Robin S. Tanke, G. Paul Torrence "Acetic Acid" in Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley-VCH, Weinheim. doi:10.1002/14356007.a01_045
- ^ Hartwig, J. F. Organotransition Metal Chemistry, from Bonding to Catalysis; University Science Books: New York, 2010. ISBN 1-891389-53-X
- ^ Zoeller, J. R.; Agreda, V. H.; Cook, S. L.; Lafferty, N. L.; Polichnowski, S. W.; Pond, D. M. (1992). "Eastman Chemical Company Acetic Anhydride Process". Catalysis Today. 13 (1): 73–91. doi:10.1016/0920-5861(92)80188-S.
- ^ Cornils, B.; Herrmann, W. A. (eds.) "Aqueous-Phase Organometallic Catalysis" VCH, Weinheim: 1998
- ^ Hartwig, John F. (2010). Organotransition metal chemistry- From bonding to Catalysis. University Science Books. ISBN 978-1-891389-53-5.
- ^ Knowles, William S. (2002). "Asymmetric Hydrogenations (Nobel Lecture)". Angewandte Chemie International Edition. 41 (12): 1999–2007. doi:10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. PMID 19746594.
- ^ H.-J.Drexler; Songlin Zhang; Ailing Sun; A. Spannenberg; A. Arrieta; A. Preetz; D. Heller (2004). "Cationic Rh-bisphosphine-diolefin complexes as precatalysts for enantioselective catalysis––what information do single crystal structures contain regarding product chirality?". Tetrahedron: Asymmetry. 15 (14): 2139–50. doi:10.1016/j.tetasy.2004.06.036.
Compounds of carbon with other elements in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Legend |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||