Congenital adrenal hyperplasia
| Congenital adrenal hyperplasia | |
|---|---|
 | |
| Congenital adrenal hyperplasia enzymes. | |
| Specialty | Endocrinology |
| Symptoms | Excessive urination of sodium, virilism, early, delayed, or absent puberty, hyperandrogenism |
| Usual onset | Before birth |
| Duration | Lifetime |
| Causes | Variants in genes responsible the enzymes required for the synthesis of cortisol in the adrenal cortex |
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis.[1][2] It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex.[3] Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids,[4][2] and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults.[5] It is one of the most common autosomal recessive disorders in humans.[6][7][8]
Types
[edit]CAH can occur in various forms. The clinical presentation of each form is different and depends to a large extent on the underlying enzyme defect, its precursor retention, and deficient products.[9] Classical forms appear in infancy, and nonclassical forms appear in late childhood. The presentation in patients with classic CAH can be further subdivided into three forms: salt-wasting, simple-virilizing, and non-classic (NC) depending on whether mineralocorticoid deficiency presents or absents, respectively.[10][11][12] This subtyping is often not clinically meaningful, though, because all patients lose salt to some degree, and clinical presentations may overlap.[13]
Classic
[edit]Salt-wasting
[edit]In 75% of cases of severe enzyme deficiency, insufficient aldosterone production can lead to salt wasting, failure to thrive, and potentially fatal hypovolemia and shock. A missed diagnosis of salt-loss CAH is related to the increased risk of early neonatal morbidity and death.[2]
Simple-virilizing
[edit]The main feature of CAH in newborn females is the abnormal development of the external genitalia, which has varying degrees of virilization. According to clinical practice guidelines, for newborns found to have bilateral inaccessible gonads, CAH evaluation should be considered. If virilizing CAH cannot be identified and treated, both boys and girls may undergo rapid postnatal growth and virilization.[2]
Nonclassic
[edit]In addition to the salt-wasting and simple-virilizing forms of CAH diagnosed in infancy, a mild or "nonclassic" form exists, which is characterized by varying degrees of postnatal androgen excess, but is sometimes asymptomatic.[14] The nonclassic form may be noticed in late childhood and may lead to signs of hyperandrogenism such as accelerated growth, acne, hirsutism, premature pubarche, menstrual irregularities,[14] and secondary polycystic ovary syndrome.[15] In adult males, early balding[14] and infertility may suggest the diagnosis. The nonclassic form is characterized by mild subclinical impairment of cortisol synthesis[14] and serum cortisol concentration is usually normal.[14]
Signs and symptoms
[edit]The symptoms of CAH vary depending upon the form of CAH and the sex of the patient. Symptoms can include:
Due to inadequate mineralocorticoids:[citation needed]
- Vomiting due to salt-wasting, leading to dehydration and death
Due to excess androgens:
- In extreme virilization, clitoromegaly (elongated clitoris) with a phallic-like structure is seen.[16][17][18]
- Ambiguous genitalia, in some infants, occurs such that initially identifying external genitalia as "male" or "female" is difficult.
- Early pubic hair and rapid growth occurs in childhood.
- Precocious puberty or failure of puberty to occur (sexual infantilism: absent or delayed puberty)
- Excessive facial hair, virilization, and/or menstrual irregularity in adolescence
- Infertility due to anovulation
- Shallow vagina[19]
Due to insufficient androgens and estrogens:[citation needed]
- Undervirilization in XY males can result in an apparent vulva.
- Ambiguous genitalia in XY males with 3β-hydroxysteroid dehydrogenase deficiency (3β-HSD2D).
- In females, hypogonadism can cause sexual infantilism or abnormal pubertal development, infertility, and other reproductive system abnormalities.
Genetics
[edit]CAH results from mutations of genes for enzymes mediating the biochemical steps of production of mineralocorticoids, glucocorticoids, or sex steroids from cholesterol by the adrenal glands (steroidogenesis).[20]
Each form of CAH is associated with a specific defective gene. The most common type (95% of cases)[2][11] involves the gene for 21-hydroxylase, which is found on 6p21.3 as part of the HLA complex; 21-hydroxylase deficiency results from a unique mutation with two highly homologous near-copies in series consisting of an active gene (CYP21A2) and an inactive pseudogene (CYP21A1P).[21][22][23] Mutant alleles result from recombination between the active and pseudogenes (gene conversion).[24] About 5% of cases of CAH are due to defects in the gene encoding 11β-hydroxylase and consequent 11β-hydroxylase deficiency. Other, more rare forms of CAH are caused by mutations in genes, including HSD3B2 (3β-hydroxysteroid dehydrogenase 2), CYP17A1 (17α-hydroxylase/17,20-lyase),[25] CYP11A1 (P450scc; cholesterol side-chain cleavage enzyme), STAR (steroidogenic acute regulatory protein; StAR), CYB5A (cytochrome b5), and CYPOR (cytochrome P450 oxidoreductase; POR).[citation needed]
Expressivity
[edit]Further variability is introduced by the degree of enzyme inefficiency produced by the specific alleles each patient has. Some alleles result in more severe degrees of enzyme inefficiency. In general, severe degrees of inefficiency produce changes in the fetus and problems in prenatal or perinatal life. Milder degrees of inefficiency are usually associated with excessive or deficient sex hormone effects in childhood or adolescence, while the mildest forms of CAH interfere with ovulation and fertility in adults.[citation needed]
Diagnosis
[edit]This section needs additional citations for verification. (October 2020) |
Clinical evaluation
[edit]Female infants with classic CAH have ambiguous genitalia due to exposure to high concentrations of androgens in utero.[26] CAH due to 21-hydroxylase deficiency is the most common cause of ambiguous genitalia in genotypically normal female infants (46XX). Less severely affected females may present with early pubarche. Young women may present with symptoms of polycystic ovarian syndrome (oligomenorrhea, polycystic ovaries, hirsutism).[medical citation needed]
Males with classic CAH generally have no signs of CAH at birth. Some may present with hyperpigmentation, due to co-secretion with melanocyte-stimulating hormone, and possible penile enlargement. Age of diagnosis of males with CAH varies and depends on the severity of aldosterone deficiency. Boys with salt-wasting disease present early with symptoms of hyponatremia and hypovolemia. Boys with non-salt-wasting disease present later with signs of virilization.[24]
In rarer forms of CAH, males are undermasculinized[27] and females generally have no signs or symptoms at birth.[medical citation needed]
Laboratory studies
[edit]Genetic analysis can be helpful to confirm a diagnosis of CAH, but it is not necessary if classic clinical and laboratory findings are present.
In classic 21-hydroxylase deficiency, laboratory studies will show:
- Hypoglycemia (due to hypocortisolism) - One of cortisol's many functions is to increase blood glucose levels. This occurs via a combination of several mechanisms, including (a) the stimulation of gluconeogesis (i.e. the creation of new glucose) in the liver, (b) the promotion of glycogenolysis (i.e. the breakdown of glycogen into glucose), and (c) the prevention of glucose leaving the bloodstream via the downregulation of GLUT-4 receptors (which normally promote movement of glucose from the bloodstream into adipose and muscle tissues). Therefore, when cortisol is deficient, these processes (effectively) occur in the reverse direction. Although there are compensatory mechanisms that mitigate the impact of hypocortisolism, they are limited in their extent and the net effect is still hypoglycemia.
- Hyponatremia (due to hypoaldosteronism) - Aldosterone is the end product of the renin-angiotensin-aldosterone system that regulates blood pressure via blood pressure surveillance in the Kidney Juxtaglomerular apparatus. Aldosterone normally functions to increase sodium retention (which brings water as well) in exchange for potassium. Thus, lack of aldosterone causes hyperkalemia and hyponatremia. In fact, this is a distinguishing point from 11-hydroxylase deficiency, in which one of the increased products is 11-deoxycorticosterone that has weak mineralocorticoid activity. In 11-hydroxylase deficiency, 11-deoxycorticosterone is produced in such excess that it acts to retain sodium at the expense of potassium. It is this reason that patients with 11-hydroxylase deficiency do not show salt wasting (although sometimes they do in infancy), and instead have hypertension/water retention and sometimes hypokalemia.
- Hyperkalemia (due to hypoaldosteronism)
- Elevated 17α-hydroxyprogesterone
Classic 21-hydroxylase deficiency typically causes 17α-hydroxyprogesterone blood levels >242 nmol/L.[medical citation needed] (For comparison, a full-term infant at three days of age should have <3 nmol/L. Many neonatal screening programs have specific reference ranges by weight and gestational age because high levels may be seen in premature infants without CAH.) Salt-wasting patients tend to have higher 17α-hydroxyprogesterone levels than non-salt-wasting patients. In mild cases, 17α-hydroxyprogesterone may not be elevated in a particular random blood sample, but it will rise during a corticotropin stimulation test.
Classification
[edit]Cortisol is an adrenal steroid hormone required for normal endocrine function. Production begins in the second month of fetal life. Poor cortisol production is a hallmark of most forms of CAH. Inefficient cortisol production results in rising levels of ACTH, because cortisol feeds back to inhibit ACTH production, so loss of cortisol results in increased ACTH.[28] This increased ACTH stimulation induces overgrowth (hyperplasia) and overactivity of the steroid-producing cells of the adrenal cortex. The defects causing adrenal hyperplasia are congenital (i.e. present at birth).
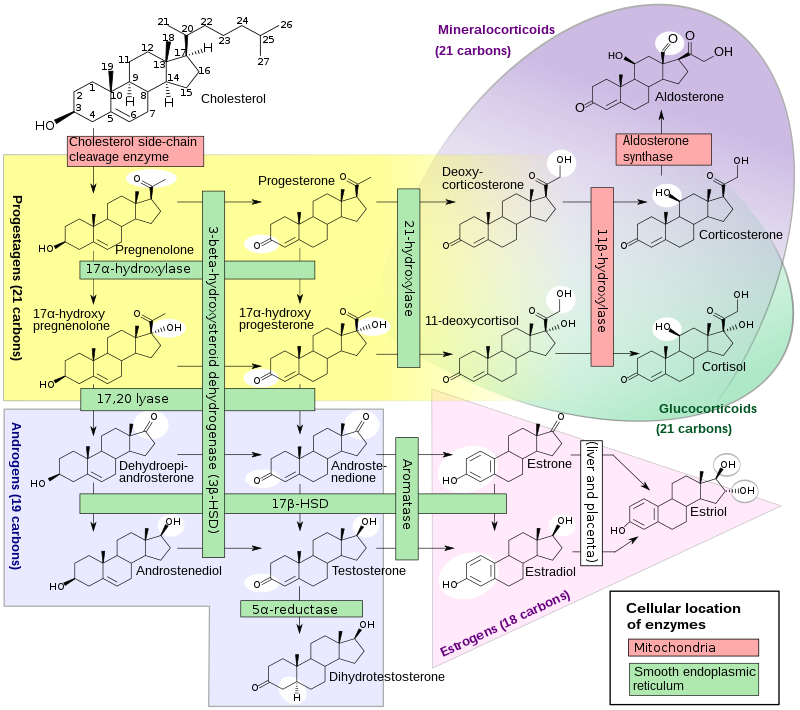
Cortisol deficiency in CAH is usually partial, and not the most serious problem for an affected person. Synthesis of cortisol shares steps with synthesis of mineralocorticoids such as aldosterone, androgens such as testosterone, and estrogens such as estradiol. The resulting excessive or deficient production of these three classes of hormones produce the most important problems for people with CAH. Specific enzyme inefficiencies are associated with characteristic patterns of over- or underproduction of mineralocorticoids or sex steroids.
Since the 1960s, most endocrinologists have referred to the forms of CAH by the traditional names in the left column, which generally correspond to the deficient enzyme activity. As exact structures and genes for the enzymes were identified in the 1980s, most of the enzymes were found to be cytochrome P450 oxidases and were renamed to reflect this. In some cases, more than one enzyme was found to participate in a reaction, and in other cases, a single enzyme mediated in more than one reaction. Variation in different tissues and mammalian species also was found.
In all its forms, congenital adrenal hyperplasia due to 21-hydroxylase deficiency accounts for about 95% of diagnosed cases of CAH.[2] Unless another specific enzyme is mentioned, "CAH" in nearly all contexts refers to 21-hydroxylase deficiency. (The terms "salt-wasting CAH", and "simple virilizing CAH" usually refer to subtypes of this condition.) CAH due to deficiencies of enzymes other than 21-hydroxylase present many of the same management challenges, as 21-hydroxylase deficiency, but some involve mineralocorticoid excess or sex steroid deficiency.
| Common medical term | % | OMIM | Enzyme(s) | Locus | Substrate(s) | Product(s) | Mineralocorticoids | Androgens |
|---|---|---|---|---|---|---|---|---|
| 21-Hydroxylase CAH | 95%[2] | 201910 | P450c21 | 6p21.3 | 17-OH-Progesterone→ Progesterone→ |
11-Deoxycortisol DOC |
↓ | ↑ |
| 11β-Hydroxylase CAH | 5% | 202010 | P450c11β | 8q21-22 | 11-Deoxycortisol→ DOC→ |
Cortisol Corticosterone |
↑ | ↑ |
| 3β-HSD CAH | Very rare | 201810 | 3βHSD2 | 1p13 | Pregnenolone→ 17-OH-Pregnenolone→ DHEA→ |
Progesterone 17-OH-Progesterone Androstenedione |
↓ | ↓ |
| 17α-Hydroxylase CAH | Very rare | 202110 | CYP17A1 | 10q24.3 | Pregnenolone→ Progesterone→ 17-OH-Pregnenolone→ |
17-OH-Pregnenolone 17-OH-Progesterone DHEA |
↑ | ↓ |
| Lipoid CAH (20,22-desmolase) |
Very rare | 201710 | StAR P450scc |
8p11.2 15q23-q24 |
Transport of cholesterol Cholesterol→ |
Into mitochondria Pregnenolone |
↓ | ↓ |
Screening
[edit]Currently, in the United States and over 40 other countries, every child born is screened for 21-hydroxylase CAH at birth. This test detects elevated levels of 17α-hydroxyprogesterone (17-OHP). Detecting high levels of 17-OHP enables early detection of CAH. Newborns detected early enough can be placed on medication and live relatively normal lives.[citation needed]
The screening process, however, is characterized by a high false-positive rate. In one study,[30] CAH screening had the lowest positive predictive value (111 true-positive cases among 20,647 abnormal screening results in a 2-year period, or 0.53%, compared with 6.36% for biotinidase deficiency, 1.84% for congenital hypo-thyroidism, 0.56% for classic galactosemia, and 2.9% for phenylketonuria). According to this estimate, 200 unaffected newborns required clinical and laboratory follow-up for every true case of CAH.[non-primary source needed]
In 2020, Wael AbdAlmageed from USC Information Sciences Institute and Mimi Kim from USC Keck School Of Medicine led a joint study in which they used deep learning technology to analyze the facial morphology and features of CAH patients compared to control. In this cross-sectional study[31] of 102 patients with CAH and 144 control participants, deep learning methods achieved a mean area under the receiver operating characteristic curve of 92% for predicting CAH from facial images. Facial features distinguished patients with CAH from controls, and analyses of facial regions found that the nose and upper face were most contributory. The findings suggest that facial morphologic features, as analyzed by deep neural network techniques, can be used as a phenotypic biomarker to predict CAH.[citation needed]
Treatment
[edit]Since the clinical manifestations of each form of CAH are unique and depend to a large extent on the underlying enzyme defects, their precursor retention and defective products, the therapeutic goal of CAH is to replenish insufficient adrenal hormones and suppress excess of precursors.[9]
Treatment of all forms of CAH may include any of:
- Supplying enough glucocorticoid to reduce hyperplasia and overproduction of androgens or mineralocorticoids[citation needed]
- Providing replacement mineralocorticoid and extra salt if the person is deficient[2]
- Providing replacement testosterone or estrogens at puberty if the person is deficient[citation needed]
- Additional treatments to optimize growth by delaying puberty or delaying bone maturation[citation needed]
If CAH is caused by the deficiency of the 21-hydroxylase enzyme, then treatment aims to normalize levels of androstenedione, but normalization of 17α-hydroxyprogesterone is a sign of overtreatment.[32] Treatment can be monitored by measuring androstenedione and 17α-hydroxyprogesterone levels in blood or saliva.[32]
Crinecerfont, an oral corticotropin-releasing factor type 1 receptor (CRF1) antagonist that reduces ACTH secretion, may allow reduction of daily glucocorticoid dosing to physiologic ranges with adequate maintenance of androstenedione control.[33][34]
Epidemiology
[edit]The incidence varies ethnically. In the United States, congenital adrenal hyperplasia in its classic form is particularly common in Native Americans and Yupik Inuit (incidence 1⁄280). Among American Caucasians, the incidence of the classic form is about 1⁄15,000).[24]
Continued treatment and wellness are enhanced by education and follow up.[35]
History
[edit]Before the twentieth century
[edit]Italian anatomist, Luigi De Crecchio (1832-1894), provided the earliest known description of a case of probable CAH.
I propose in this narrative that it is sometimes extremely difficult and even impossible to determine sex during life. In one of the anatomical theaters of the hospital..., there arrived toward the end of January a cadaver which in life was the body of a certain Joseph Marzo... The general physiognomy was decidedly male in all respects. There were no feminine curves to the body. There was a heavy beard. There was some delicacy of structure with muscles that were not very well developed... The distribution of pubic hair was typical of the male. Perhaps the lower extremities were somewhat delicate, resembling the female, and were covered with hair... The penis was curved posteriorly and measured 6 cm, or with stretching, 10 cm. The corona was 3 cm long and 8 cm in circumference. There was an ample prepuce. There was a first grade hypospadias... There were two folds of skin coming from the top of the penis and encircling it on either side. These were somewhat loose and resembled labia majora.
De Crecchio then described the internal organs, which included a normal vagina, uterus, fallopian tubes, and ovaries.
It was of the greatest importance to determine the habits, tendencies, passions, and general character of this individual... I was determined to get as complete a story as possible, determined to get at the base of the facts and to avoid undue exaggeration which was rampant in the conversation of many of the people present at the time of the dissection.
He interviewed many people and satisfied himself that Joseph Marzo "conducted himself within the sexual area exclusively as a male", even to the point of contracting the "French disease" (syphilis) on two occasions. The cause of death was another in a series of episodes of vomiting and diarrhea.[36]
This account was translated by Alfred Bongiovanni from De Crecchio ("Sopra un caso di apparenzi virili in una donna". Morgagni 7:154–188, 1865) in 1963 for an article in The New England Journal of Medicine.[citation needed]
Twentieth and twenty-first centuries
[edit]The association of excessive sex steroid effects with diseases of the adrenal cortex have been recognized for over a century. The term "adrenogenital syndrome" was applied to both sex-steroid producing tumors and severe forms of CAH for much of the 20th century, before some of the forms of CAH were understood. Congenital adrenal hyperplasia, which also dates to the first half of the century, has become the preferred term to reduce ambiguity and to emphasize the underlying pathophysiology of the disorders.[citation needed]
Much modern understanding and treatment of CAH comes from research conducted at Johns Hopkins Medical School in Baltimore in the middle of the 20th century. Lawson Wilkins, "founder" of pediatric endocrinology, worked out the apparently paradoxical pathophysiology: that hyperplasia and overproduction of adrenal androgens resulted from impaired capacity for making cortisol. He reported use of adrenal cortical extracts to treat children with CAH in 1950. Genital reconstructive surgery was also pioneered at Hopkins. After application of karyotyping to CAH and other intersex disorders in the 1950s, John Money, JL Hampson, and JG Hampson persuaded both the scientific community and the public [citation needed] that sex assignment should not be based on any single biological criterion, and gender identity was largely learned and has no simple relationship with chromosomes or hormones. See Intersex for a fuller history, including recent controversies over reconstructive surgery.[citation needed]
Hydrocortisone, fludrocortisone, and prednisone were available by the late 1950s. By 1980, all of the relevant steroids could be measured in blood by reference laboratories for patient care. By 1990, nearly all specific genes and enzymes had been identified. The last decade, though, has seen a number of new developments, discussed more extensively in congenital adrenal hyperplasia due to 21-hydroxylase deficiency:[citation needed]
- Debate over the value of genital reconstructive surgery and changing standards
- Debate over sex assignment of severely virilized XX infants
- New treatments to improve height outcomes
- Newborn screening programs to detect CAH at birth
- Increasing attempts to treat CAH before birth
Society and culture
[edit]People with CAH
[edit]Notable people with CAH include:
- Jeff Cagandahan is a Filipino who successfully appealed for a change of name and gender on his birth certificate.[37]
- Lisa Lee Dark[38]
- Betsy Driver[39]
- Casimir Pulaski, hypothesized based on examination of remains[40]
See also
[edit]- Disorders of sex development
- Inborn errors of steroid metabolism
- Intersex
- List of vaginal anomalies
- 5α-reductase 2 deficiency
- Androgen insensitivity syndrome
References
[edit]- ^ El-Maouche D, Arlt W, Merke DP (November 2017). "Congenital adrenal hyperplasia" (PDF). Lancet. 390 (10108): 2194–2210. doi:10.1016/S0140-6736(17)31431-9. PMID 28576284. S2CID 13737960.
- ^ a b c d e f g h Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC (2018). "Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline". The Journal of Clinical Endocrinology and Metabolism. 103 (11): 4043–4088. doi:10.1210/jc.2018-01865. PMC 6456929. PMID 30272171.
- ^ Speiser PW, White PC (August 2003). "Congenital adrenal hyperplasia". The New England Journal of Medicine. 349 (8): 776–88. doi:10.1056/NEJMra021561. PMID 12930931.
- ^ La, Betty; Tung, Celestine; Choi, Eugene A.; Nguyen, Ha (1 November 2021). "A Gigantic Uterine Leiomyoma and Big Bilateral Adrenal Myelolipomas as a Result of Untreated Congenital Adrenal Hyperplasia". AACE Clinical Case Reports. 7 (6): 342–345. doi:10.1016/j.aace.2021.05.002. PMC 8573279. PMID 34765728.
- ^ Aubrey Milunsky; Jeff Milunsky (29 January 2010). Genetic Disorders and the Fetus: Diagnosis, Prevention and Treatment. John Wiley and Sons. pp. 600–. ISBN 978-1-4051-9087-9. Retrieved 14 June 2010.
- ^ Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI (July 1985). "High frequency of nonclassical steroid 21-hydroxylase deficiency". American Journal of Human Genetics. 37 (4): 650–67. PMC 1684620. PMID 9556656.
- ^ Krone N, Arlt W (April 2009). "Genetics of congenital adrenal hyperplasia". Best Practice & Research. Clinical Endocrinology & Metabolism. 23 (2): 181–92. doi:10.1016/j.beem.2008.10.014. PMC 5576025. PMID 19500762.
- ^ Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, Auchus RJ (May 2016). "Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency". European Journal of Endocrinology. 174 (5): 601–9. doi:10.1530/EJE-15-1181. PMC 4874183. PMID 26865584.
- ^ a b Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP, New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, Yuen T, Khattab A (15 March 2017). Congenital Adrenal Hyperplasia. MDText.com, Inc. PMID 25905188.
- ^ Speiser, Phyllis W.; Arlt, Wiebke; Auchus, Richard J.; Baskin, Laurence S.; Conway, Gerard S.; Merke, Deborah P.; Meyer-Bahlburg, Heino F. L.; Miller, Walter L.; Murad, M. Hassan; Oberfield, Sharon E.; White, Perrin C. (1 November 2018). "Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline". The Journal of Clinical Endocrinology and Metabolism. 103 (11): 4043–4088. doi:10.1210/jc.2018-01865. ISSN 1945-7197. PMC 6456929. PMID 30272171.
- ^ a b Nordenström, Anna; Lajic, Svetlana; Falhammar, Henrik (8 July 2022). "Long-Term Outcomes of Congenital Adrenal Hyperplasia". Endocrinology and Metabolism. 37 (4): 587–598. doi:10.3803/EnM.2022.1528. ISSN 2093-596X. PMC 9449109. PMID 35799332.
- ^ Dauber A, Kellogg M, Majzoub JA (2010). "Monitoring of therapy in congenital adrenal hyperplasia". Clinical Chemistry. 56 (8): 1245–51. doi:10.1373/clinchem.2010.146035. PMID 20558634.
- ^ Merke DP, Auchus RJ (September 2020). "Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency". The New England Journal of Medicine. 383 (13): 1248–1261. doi:10.1056/NEJMra1909786. PMID 32966723. S2CID 221884108.
- ^ a b c d e Adriaansen, Bas P. H.; Schröder, Mariska A. M.; Span, Paul N.; Sweep, Fred C. G. J.; van Herwaarden, Antonius E.; Claahsen-van der Grinten, Hedi L. (12 December 2022). "Challenges in treatment of patients with non-classic congenital adrenal hyperplasia". Frontiers in Endocrinology. 13. doi:10.3389/fendo.2022.1064024. PMC 9791115. PMID 36578966.
- ^ Yau, M.; et al. (2000). Congenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment. MDText.com. PMID 25905311.
- ^ Philadelphia, The Children's Hospital of (19 November 2019). "Classic congenital Adrenal Hyperplasia Diagnosed in the Newborn Period". www.chop.edu. Retrieved 5 September 2020.
- ^ New, Maria; Yau, Mabel; Lekarev, Oksana; Lin-Su, Karen; Parsa, Alan; Pina, Christian; Yuen, Tony; Khattab, Ahmed (15 March 2017). "Figure 2, [Different degrees of virilization according...]". www.ncbi.nlm.nih.gov. Retrieved 5 September 2020.
- ^ "Genital Birth Defects — Children's Health Issues". Merck Manuals Consumer Version. Retrieved 5 September 2020.
- ^ Richard D. McAnulty, M. Michele Burnette (2006) Sex and sexuality, Volume 1, Greenwood Publishing Group, p.165
- ^ David A. Warrell (2005). Oxford textbook of medicine: Sections 18-33. Oxford University Press. pp. 261–. ISBN 978-0-19-856978-7. Retrieved 14 June 2010.
- ^ Gidlöf, Sebastian; Falhammar, Henrik; Thilén, Astrid; Döbeln, Ulrika von; Ritzén, Martin; Wedell, Anna; Nordenström, Anna (1 September 2013). "One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study". The Lancet Diabetes & Endocrinology. 1 (1): 35–42. doi:10.1016/S2213-8587(13)70007-X. ISSN 2213-8587. PMID 24622265.
- ^ Arlt, Wiebke; Willis, Debbie S.; Wild, Sarah H.; Krone, Nils; Doherty, Emma J.; Hahner, Stefanie; Han, Thang S.; Carroll, Paul V.; Conway, Gerry S.; Rees, D. Aled; Stimson, Roland H.; Walker, Brian R.; Connell, John M. C.; Ross, Richard J. (18 August 2010). "Health Status of Adults with Congenital Adrenal Hyperplasia: A Cohort Study of 203 Patients". The Journal of Clinical Endocrinology & Metabolism. 95 (11): 5110–5121. doi:10.1210/jc.2010-0917. ISSN 0021-972X. PMC 3066446. PMID 20719839.
- ^ Strömer, H.; Cittadini, A.; Douglas, P. S.; Morgan, J. P. (12 January 2022). "Exogenously administered growth hormone and insulin-like growth factor-I alter intracellular Ca2+ handling and enhance cardiac performance. In vitro evaluation in the isolated isovolumic buffer-perfused rat heart". Circulation Research. 79 (2): 227–236. doi:10.1161/01.res.79.2.227. ISSN 0009-7330. PMID 8755999.
- ^ a b c Mais, Daniel D. (2008). Quick compendium of clinical pathology (2nd ed.). Chicago: ASCP Press. ISBN 978-0891895671.
- ^ Miller WL (January 2012). "The syndrome of 17,20 lyase deficiency". The Journal of Clinical Endocrinology and Metabolism. 97 (1): 59–67. doi:10.1210/jc.2011-2161. PMC 3251937. PMID 22072737.
- ^ White, P. C.; Speiser, P. W. (1 June 2000). "Congenital adrenal hyperplasia due to 21-hydroxylase deficiency". Endocrine Reviews. 21 (3): 245–291. doi:10.1210/edrv.21.3.0398. ISSN 0163-769X. PMID 10857554.
- ^ "3-beta-hydroxysteroid dehydrogenase deficiency". MedlinePlus. Archived from the original on 28 September 2020. Retrieved 2 June 2021.
- ^ Kumar, Vinay; Abbas, Abul K.; Aster, Jon C. (2014). Robbins and Cotran pathologic basis of disease. Kumar, Vinay, 1944-, Abbas, Abul K.,, Aster, Jon C.,, Perkins, James A. (Ninth ed.). Philadelphia, PA. p. 1128. ISBN 9781455726134. OCLC 879416939.
{{cite book}}: CS1 maint: location missing publisher (link) - ^ Häggström, Mikael; Richfield, David (2014). "Diagram of the pathways of human steroidogenesis". WikiJournal of Medicine. 1 (1). doi:10.15347/wjm/2014.005. ISSN 2002-4436.
- ^ Kenneth A. Pass; Eurico Carmago Neto (2005). Update: Newborn Screening for Endocrinopathies (PDF). pp. 831–834. Archived from the original (PDF) on 1 January 2014. Retrieved 12 December 2013.
- ^ AbdAlmageed, Wael; Mirzaalian, Hengameh; Guo, Xiao; Randolph, Linda M.; Tanawattanacharoen, Veeraya K.; Geffner, Mitchell E.; Ross, Heather M.; Kim, Mimi S. (18 November 2020). "Assessment of Facial Morphologic Features in Patients With Congenital Adrenal Hyperplasia Using Deep Learning". JAMA Network Open. 3 (11): e2022199. doi:10.1001/jamanetworkopen.2020.22199. ISSN 2574-3805. PMC 7675110. PMID 33206189.
- ^ a b Adriaansen, Bas P. H.; Kamphuis, Johannes S.; Schröder, Mariska A. M.; Olthaar, André J.; Bock, Carina; Brandt, André; Stikkelbroeck, Nike M. M. L.; Lentjes, Eef G. W. M.; Span, Paul N.; Sweep, Fred C. G. J.; Claahsen-van der Grinten, Hedi L.; van Herwaarden, Antonius E. (2022). "Diurnal salivary androstenedione and 17-hydroxyprogesterone levels in healthy volunteers for monitoring treatment efficacy of patients with congenital adrenal hyperplasia". Clinical Endocrinology. 97 (1): 36–42. doi:10.1111/cen.14690. ISSN 0300-0664. PMC 9542109. PMID 35150157.
- ^ Auchus, Richard J.; Hamidi, Oksana; Pivonello, Rosario; Bancos, Irina; Russo, Gianni; Witchel, Selma F.; Isidori, Andrea M.; Rodien, Patrice; Srirangalingam, Umasuthan; Kiefer, Florian W.; Falhammar, Henrik; Merke, Deborah P.; Reisch, Nicole; Sarafoglou, Kyriakie; Cutler, Gordon B. (June 2024). "Phase 3 Trial of Crinecerfont in Adult Congenital Adrenal Hyperplasia". New England Journal of Medicine. doi:10.1056/NEJMoa2404656. ISSN 0028-4793. PMC 11309900. PMID 38828955.
- ^ Sarafoglou, Kyriakie; Kim, Mimi S.; Lodish, Maya; Felner, Eric I.; Martinerie, Laetitia; Nokoff, Natalie J.; Clemente, María; Fechner, Patricia Y.; Vogiatzi, Maria G.; Speiser, Phyllis W.; Auchus, Richard J.; Rosales, Gelliza B.G.; Roberts, Eiry; Jeha, George S.; Farber, Robert H. (2 June 2024). "Phase 3 Trial of Crinecerfont in Pediatric Congenital Adrenal Hyperplasia". New England Journal of Medicine. doi:10.1056/NEJMoa2404655. ISSN 0028-4793. PMID 38828945.
- ^ Kruse, B.; Riepe, F. G.; Krone, N.; Bosinski, H. a. G.; Kloehn, S.; Partsch, C. J.; Sippell, W. G.; Mönig, H. (July 2004). "Congenital adrenal hyperplasia — how to improve the transition from adolescence to adult life". Experimental and Clinical Endocrinology & Diabetes. 112 (7): 343–355. doi:10.1055/s-2004-821013. ISSN 0947-7349. PMID 15239019. S2CID 260138410.
- ^ Bongiovanni AM, Root AW (1963). "The Adrenogenital Syndrome". The New England Journal of Medicine. 268 (23): 1283–9 contd. doi:10.1056/NEJM196306062682308. PMID 13968788.
- ^ International Commission of Jurists (2017). "Republic of the Philippines v. Jennifer Cagandahan, Supreme Court of the Philippines, Second Division (12 September 2008)". International Commission of Jurists. Retrieved 19 March 2021.
- ^ "BBC Radio 4 – Changing Sex". Retrieved 6 August 2008.
- ^ "Mayor Betsy Driver is Promoting Intersex Visibility Through Activism and Politics". Yahoo. 23 August 2019. Archived from the original on 22 September 2021. Retrieved 10 September 2019.
- ^ Schoenberg, Nara (3 April 2019). "'It's a woman. It's not Pulaski.': New documentary argues Revolutionary War hero was intersex". chicagotribune.com. Archived from the original on 2 November 2019. Retrieved 28 May 2020.
Further reading
[edit]- Han, Thang S.; Walker, Brian R.; Arlt, Wiebke; Ross, Richard J. (17 December 2013). "Treatment and health outcomes in adults with congenital adrenal hyperplasia". Nature Reviews Endocrinology. 10 (2): 115–124. doi:10.1038/nrendo.2013.239. PMID 24342885. S2CID 6090764Figure 2: The adrenal steroidogenesis pathway.
{{cite journal}}: CS1 maint: postscript (link)