Idiopathische Lungenfibrose
Die idiopathische Lungenfibrose oder idiopathische pulmonale Fibrose (IPF) ist eine sehr schwerwiegende chronische Erkrankung mit oft tödlichem Ausgang, die durch eine stetige Abnahme der Lungenfunktion gekennzeichnet ist.[1][2] Der Begriff Lungenfibrose steht für eine Vernarbung des Lungengewebes, die zu einer ständig zunehmenden Dyspnoe (Atemnot) führt. Die Fibrose hat meistens eine schlechte Prognose.[1][2][3] Der Begriff „idiopathisch“ wird verwendet, da die Ursache der Lungenfibrose noch nicht bekannt ist.[1]
IPF tritt meistens im Erwachsenenalter zwischen 50 und 70 Jahren auf, vor allem bei aktiven oder ehemaligen Rauchern; Männer sind häufiger betroffen als Frauen.[1][4]
IPF gehört zu einer großen Gruppe von etwa 200 Lungenerkrankungen, die als interstitielle Lungenerkrankungen (englisch Interstitial Lung Disease oder ILD) bezeichnet werden und einen Befall des Lungeninterstitiums aufweisen.[2] Das Interstitium, also das Bindegewebe zwischen den Lungenbläschen (Alveolen), ist hauptsächlich befallen. Dennoch befallen diese Erkrankungen häufig nicht nur das Interstitium, sondern auch Alveolen, periphere Atemwege und Gefäße.[2] Das Lungengewebe von Personen mit IPF weist ein charakteristisches histopathologisches Muster auf, das als gewöhnliche interstitielle Pneumonie (englisch Usual Interstitial Pneumonia oder UIP) bezeichnet wird. UIP ist die histologisch bzw. detailradiologische Entsprechung der IPF.[1][4]
2011 wurden neue Richtlinien für Diagnose und Management der IPF veröffentlicht.[1] Eine deutschsprachige Fassung der internationalen Richtlinien aus dem Jahr 2013 basiert auf einer Initiative deutscher Experten unter der Schirmherrschaft der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin (DGP) und auf den Ergebnissen einer Konsensuskonferenz.[5]
Die Diagnose der IPF setzt den Ausschluss anderer Formen einer interstitiellen Pneumonie voraus, einschließlich anderer idiopathischer interstitieller Pneumonien und interstitieller Lungenerkrankungen (ILD) im Zusammenhang mit Umweltbelastungen, Medikamenten oder systemischen Erkrankungen.[5]
Klassifikation
[Bearbeiten | Quelltext bearbeiten]| Klassifikation nach ICD-10 | |
|---|---|
| J84.112 | Idiopathische Lungenfibrose |
| ICD-10 online (WHO-Version 2019) | |
Die IPF gehört zu einer großen Gruppe von mehr als 200 Lungenerkrankungen, die als interstitielle Lungenerkrankungen (ILD) bezeichnet werden und durch einen Befall des Lungeninterstitiums,[2] also des Bindegewebes zwischen den Lungenbläschen, gekennzeichnet sind. Die IPF ist eine Form der idiopathischen interstitiellen Pneumonie (IIP), die wiederum eine Art ILD ist, auch bekannt als diffuse parenchymale Lungenerkrankung (englisch Diffuse Parenchymal Lung Disease oder DPLD).
Die Klassifikation von IIPs der American Thoracic Society/European Respiratory Society (ATS/ERS) aus dem Jahr 2002 wurde 2013 aktualisiert.[2]
Diese neue Klassifikation umfasst drei Hauptkategorien an IIPs: häufig auftretende IIPs, selten auftretende IIPs und nicht klassifizierbare IIPs. Zu den häufig auftretenden IIPs zählen chronisch fibrosierende IPs (darunter IPF und nichtspezifische interstitielle Pneumonie [NSIP]), rauchbedingte IPs (respiratorische Bronchiolitis mit interstitieller Lungenerkrankung [RB-ILD] und desquamative interstitielle Pneumonie [DIP]) sowie akute/subakute IPs (kryptogene organisierende Pneumonie [COP] und akute interstitielle Pneumonie [AIP]).[2]
Zur Diagnose von IIPs müssen bekannte Ursachen von ILD ausgeschlossen werden. Beispiele für ILDs mit bekannten Ursachen sind unter anderem hypersensitive Pneumonitis, Langerhans-Zell-Histiozytose, Asbestose und Kollagen-Gefäßkrankheit. Diese Krankheiten betreffen jedoch häufig nicht nur das Interstitium, sondern auch die peripheren Atemwege und Gefäße.[2]
Epidemiologie
[Bearbeiten | Quelltext bearbeiten]Groß angelegte Studien zur Inzidenz und Prävalenz der IPF fehlen.[5]
Obwohl sehr selten, so ist die IPF doch die häufigste Form von IIP.[2] Basierend auf einer Analyse US-amerikanischer Forderungen gegenüber Krankenkassen wird die Verbreitung von IPF auf 14,0–42,7 Fälle pro 100.000 Einwohner geschätzt. Die breite Streuung ergibt sich aufgrund der Definitionen, die in den jeweiligen Fällen verwendet wurden.[4][5] IPF tritt häufiger bei Männern als bei Frauen auf und wird für gewöhnlich bei Personen über 50 Jahren diagnostiziert.[1]
Die Inzidenz der IPF ist schwer zu bestimmen, da einheitliche diagnostische Kriterien nur inkonsequent angewendet wurden.[1][5] In den 27 EU-Mitgliedstaaten wird die Inzidenz durch verschiedene Quellen auf 4,6–7,4 Personen pro 100.000 Einwohner geschätzt,[6][7] woraus man schließen kann, dass jedes Jahr etwa 30.000–35.000 neue Patienten mit IPF diagnostiziert werden.[4][8]
Eine kürzlich am Universitätskrankenhaus Århus (Dänemark) durchgeführte monozentrische, retrospektive Beobachtungs- und Kohortenstudie mit Patienten, bei denen zwischen 2003 und 2009 ILD diagnostiziert wurde, ergab eine Inzidenz von 4,1 pro 100.000 Einwohnern/Jahr für ILD. Die häufigste Diagnose lautete IPF (28 %), gefolgt von ILD in Zusammenhang mit Bindegewebserkrankungen (14 %), Hypersensitivitäts-Pneumonitis (7 %) und nichtspezifischer interstitieller Pneumonie (NSIP) (7 %). Die Inzidenz von IPF betrug 1,3 pro 100.000 Einwohner/Jahr.[9]
Aufgrund einer ungleichmäßigen Verbreitung der Krankheit in europäischen Ländern sollten epidemiologische Daten mittels eines europaweiten ILD- und IPF-Registers aktualisiert werden. In Deutschland wurde von 2012 bis 2020 das akademische INSIGHTS-IPF Register (Investigating Significant Health Trends in IPF, Studienleiter Prof. Jürgen Behr) geführt, in dem 1232 Patienten dokumentiert werden. Die Ergebnisse zeigen, dass IPF-Patienten in der klinischen Routine kränker sind und eine schlechtere Lebensqualität haben als in den kontrollierten Studien, und die Behandlungsansätze sehr unterschiedlich ausfallen.[10][11][12][13] Das Register wird seit dem Jahr 2021 ergänzt durch das INSIGHTS-ILD[14] Register derselben Studiengruppe, das progressiv fibrosierende interstielle Lungenerkrankungen umfassend dokumentiert.[15]
Ursachen/Risikofaktoren für IPF
[Bearbeiten | Quelltext bearbeiten]IPF oder idiopathische Lungenfibrose ist per definitionem idiopathisch (d. h., es liegt keine bekannte Ursache vor), einige Umwelt- und Expositionsfaktoren können jedoch das Risiko einer Erkrankung mit IPF erhöhen.[16] Rauchen gilt als einer der zentralen Risikofaktoren für IPF, da es das IPF-Risiko in etwa verdoppelt.[16]
Weitere Umwelteinflüsse und eine Exposition am Arbeitsplatz wie z. B. gegenüber Metallstaub, Holzstaub, Kohlenstaub, Steinstaub oder Siliziumdioxid sowie Tätigkeiten im Bereich der Landwirtschaft/Viehzucht erhöhen ebenfalls erwiesenermaßen das Risiko von IPF. Es liegen Nachweise dafür vor, dass Virusinfektionen mit idiopathischer Lungenfibrose und anderen fibrosierenden Lungenerkrankungen in Zusammenhang stehen könnten.[17]
Ätiologie und Pathobiologie
[Bearbeiten | Quelltext bearbeiten]Trotz intensiver Forschung bleibt die Ursache der IPF unbekannt.[1] Die bei IPF auftretende Fibrose wird mit Rauchen von Zigaretten, Umweltfaktoren (z. B. dem berufsbedingten Kontakt mit Gasen, Rauch, Chemikalien oder Staub), anderen Krankheiten wie zum Beispiel Refluxösophagitis (englisch Gastroesophageal Reflux Disease oder GERD) oder mit genetischer Prädisposition (familiäre IPF) in Verbindung gebracht.
Jedoch sind keine dieser Faktoren bei allen IPF-Patienten nachweisbar, so dass sie keine zufriedenstellende Erklärung für die Krankheit liefern.[1][18]
Es wird davon ausgegangen, dass die IPF das Ergebnis eines aberranten Wundheilungsprozesses ist, der mit abnormaler und exzessiver Kollagenablagerung (Fibrose) im Lungeninterstitium sowie mit einer minimalen Entzündung einhergeht.[19]
Man vermutet, dass bei IPF eine initiale oder wiederholte Verletzung von Lungenzellen, den sogenannten alveolären Epithelzellen oder Pneumozyten Typ I und Typ II (englisch Alveolar Epithelial Cells (AECs), die den Großteil der Oberfläche des Lungenbläschens auskleiden, zugrunde liegt.[20] Wenn Pneumozyten Typ I beschädigt werden oder untergehen, proliferieren vermutlich Pneumozyten Typ II, um die freiliegende Basalmembran zu bedecken.
Bei einem normalen Reparaturvorgang sterben die hyperplastischen Pneumozyten Typ II ab, während sich die übrigen Zellen ausbreiten und einen Differenzierungsprozess zu Pneumozyten Typ I durchlaufen.
Unter pathologischen Bedingungen und in Anwesenheit des TGF-β (englisch Transforming Growth Factor Beta) akkumulieren Fibroblasten in der geschädigten Region und differenzieren zu Myofibroblasten, die Kollagen und andere Proteine absondern.[20] In der Vergangenheit vertrat man die Annahme, dass eine Entzündung das erste Ereignis darstellt, das eine Vernarbung des Lungengewebes einleitet. Neuesten Erkenntnissen zufolge geht jedoch die Entwicklung fibroblastischer Foki der Ansammlung von Entzündungszellen und der daraus resultierenden Ablagerung von Kollagen voraus.[21]

Dieses pathogenetische Modell wird indirekt durch die klinischen Merkmale der IPF unterstützt, einschließlich des schleichenden Krankheitsbeginns, der Progression über mehrere Jahre, den relativ seltenen akuten Exazerbationen und des Nichtansprechens auf Immunsuppressiva.[19][22] Therapien, die auf die Bekämpfung der Fibroblasten-Aktivierung oder auf die Synthese extrazellulärer Matrix ausgerichtet sind, befinden sich zurzeit in einer frühen Testphase oder werden zur Weiterentwicklung in Betracht gezogen.
Die familiäre IPF macht weniger als 5 % aller Patienten mit IPF aus und lässt sich klinisch und histologisch nicht von der sporadischen IPF unterscheiden.[1] Genetische Assoziationen umfassen Mutationen der Surfactantproteine A1, A2, C (SFTPA1, SFTPA2B) und der Mucine (MUC5B).[23] Ein interessanter Aspekt der MUC5B-Variante ist ihre Erkennungsrate, da sie bei etwa 20 % der Patienten nord- und westeuropäischer Abstammung und bei 19 % der Population der Framingham-Herz-Studie festgestellt wird.[24] Mutationen menschlicher Telomerase-Gene werden ebenfalls mit familiärer Lungenfibrose in Verbindung gebracht, bei manchen Patienten auch mit sporadischer IPF (TERT, TERC).[23] Vor Kurzem wurde eine X-chromosomale Mutation in einem dritten Telomerase-assoziierten Gen, Dyskerin (DKC1), in einer Familie mit IPF beschrieben.[25]
Diagnose
[Bearbeiten | Quelltext bearbeiten]Eine möglichst frühe Diagnose der IPF ist die Voraussetzung für einen früheren Behandlungsbeginn und somit für eine mögliche Verbesserung des klinischen Langzeitergebnisses dieser progressiven und letztendlich tödlichen Krankheit.[1] Wenn Verdacht auf IPF besteht, kann die Diagnose eine Herausforderung darstellen; es hat sich jedoch gezeigt, dass ein multidisziplinärer Ansatz unter Hinzuziehung von Experten auf dem Gebiet interstitieller Lungenerkrankungen aus den Fachbereichen Pulmologie, Radiologie und Pathologie die Treffsicherheit der IPF-Diagnose verbessert.[1][26][27]
Ein multidisziplinäres Konsensus-Statement zu idiopathischen interstitiellen Pneumonien der American Thoracic Society (ATS) und der European Respiratory Society (ERS) aus dem Jahr 2000 schlug verschiedene Major- und Minor-Kriterien vor, die der IPF-Diagnosestellung dienen.[1] 2011 hat die ATS und ERS zusammen mit der Japanese Respiratory Society (JRS) und der Latin American Thoracic Association (ALAT) neue, vereinfachte und aktualisierte Kriterien für Diagnose und Management der IPF veröffentlicht.[1] Heute erfordert eine IPF-Diagnose:
- den Ausschluss bekannter Ursachen einer ILD, wie z. B.: häusliche oder durch den Arbeitsplatz bedingte Umweltfaktoren, Bindegewebserkrankungen oder Medikamenteneinwirkung/-Toxizität;
- das Auftreten eines typischen radiologischen UIP-Musters im HRCT.
In der Regel lässt sich die IPF durch ein HRCT allein diagnostizieren, wodurch eine chirurgische Lungenbiopsie vermieden werden kann.[1][2]
In der klinischen Praxis kann die Erkennung der IPF eine große Herausforderung darstellen, da die Symptome oft den Symptomen häufiger auftretender Krankheiten ähneln, wie etwa Asthma, chronisch obstruktiver Lungenkrankheit (englisch Chronic Obstructive Pulmonary Disease oder COPD) und Herzinsuffizienz.[28] Das Hauptproblem, das sich Ärzten stellt, ist die Frage, ob die vorliegende Anamnese, die Symptome (oder Anzeichen), die radiologischen Befunde und die Lungenfunktionstests alle einer IPF-Diagnose entsprechen, oder ob die Befunde das Ergebnis eines anderen Krankheitsprozesses sind. Es ist schon lange bekannt, dass die ILD, hervorgerufen durch Asbest-Exposition, Medikamente (z. B. Chemotherapeutika oder Nitrofurantoin), rheumatoide Arthritis und Sklerodermie nur schwer von der IPF unterschieden werden kann. Andere differentialdiagnostische Überlegungen müssen eine interstitielle Lungenerkrankung in Verbindung mit Mischkollagenose, fortgeschrittener Sarkoidose, chronischer hypersensitiver Pneumonitis, Langerhans-Zell-Histiozytose und strahlungsinduzierter Fibrose umfassen.[1][2]
Klinische Merkmale
[Bearbeiten | Quelltext bearbeiten]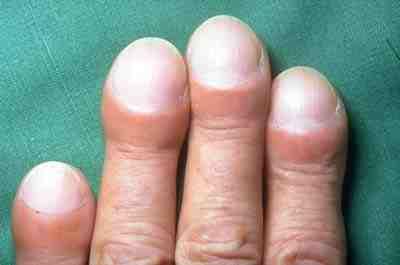
Bei vielen Patienten sind Symptome bereits lange vor der Diagnosestellung evident.[3] Zu den häufigsten klinischen Merkmalen der IPF gehören:[1][2][29]
- Alter über 50 Jahre
- trockener, nicht-produktiver Husten bei Belastung
- fortschreitende Belastungsdyspnoe (Atemnot bei Belastung)
- trockenes, inspiratorisches bi-basiläres Klettverschluss-artiges Rasselgeräusch bei der Auskultation mit dem Stethoskop (ein inspiratorisches Knisterrasseln in der Lunge, das wie ein Klettverschluss klingt, der langsam auseinandergezogen wird)[1][5][30]
- Trommelschlegelbildung, eine Verformung der Finger- und Zehenspitzen (siehe Abbildung)
- anormale Ergebnisse beim Lungenfunktionstest, mit Nachweis eines eingeschränkten und beeinträchtigten Gasaustausches.
Diese Merkmale lassen sich zum Teil auf chronischen Sauerstoffmangel im Blut zurückführen. Sie können jedoch bei vielen Lungenerkrankungen auftreten und sind daher nicht IPF-spezifisch. IPF sollte jedoch bei allen Patienten mit nicht geklärter Belastungsdyspnoe in Erwägung gezogen werden, bei denen Symptome wie Husten, bi-basiläres inspiratorisches Knisterrasseln oder Trommelschlegelfinger vorliegen.[1]
Die Feststellung von Knisterrasseln bei der Lungenauskultation trägt zur früheren IPF-Diagnose bei. Feine Knistergeräusche können von Ärzten leicht erkannt werden und sind charakteristisch für die IPF.[31]
Wenn bei einem Patienten über 60 Jahre bilaterale Knistergeräusche während der gesamten Inspiration zu hören sind, wenn sie auch nach einigen tiefen Atemzügen weiterhin bestehen und zu unterschiedlichen Zeitpunkten, die mehrere Wochen auseinander liegen können, festgestellt werden, so sollte man an eine IPF denken und ein HRCT in Erwägung ziehen, das empfindlicher als eine Röntgenübersicht des Thorax ist.[30] Da Knistergeräusche nicht IPF-spezifisch sind, müssen sie zur Einleitung eines sorgfältigen Diagnoseprozesses führen.[1]
Das Erkennen dieser Symptome erfordert angemessene weiterführende Untersuchungen zur Diagnose der IPF.
Radiologie
[Bearbeiten | Quelltext bearbeiten]Röntgenübersichten des Thorax sind sehr nützlich bei der Verlaufsbeobachtung von IPF-Patienten. Das konventionelle Übersichtsbild führt leider zu keiner endgültigen Diagnose, kann aber ein verringertes Lungenvolumen anzeigen, typischerweise mit markanten retikulären interstitiellen Zeichnungsvermehrungen nahe der Lungenbasis.[1]

Die radiologische Untersuchung mittels HRCT ist ein grundlegendes Element der IPF-Diagnose. Ein HRCT wird mit einem konventionellen Computertomographen ohne Injektion eines Kontrastmittels durchgeführt. Die Schnittbilder zur Auswertung sind sehr dünn (1–2 mm).
Ein typisches Thorax-HRCT bei IPF weist fibrotische Veränderungen beider Lungenflügel auf, vor allem im Bereich der Basis und der Peripherie. Gemäß der gemeinsamen ATS/ERS/JRS/ALAT-Richtlinien von 2011 ist das HRCT ein maßgeblicher Bestandteil im diagnostischen Prozess der IPF, da es eine UIP bei Vorliegen folgender Merkmale identifizieren kann:[1]
- Retikuläre Verschattungen, die oft mit Traktionsbronchiektasie einhergehen
- Honigwabenmuster, gekennzeichnet durch Ansammlungen zystischer Lufträume, die meistens einen vergleichbaren Durchmesser haben (3–10 mm), gelegentlich aber auch größer sind. Sie liegen für gewöhnlich subpleural und sind durch klar abgegrenzte Wände und das Auftreten in mindestens zwei Reihen gekennzeichnet. Eine Reihe von Zysten reicht nicht aus, um als Honigwabenmuster bezeichnet zu werden.
- Milchglasverschattungen sind häufig, doch weniger ausgedehnt als die Netzstruktur.
- Die Verteilung ist typischerweise basal und peripher, jedoch häufig auch fleckförmig.
Histologie
[Bearbeiten | Quelltext bearbeiten]Gemäß den 2011 aktualisierten Richtlinien ist für eine verlässliche Diagnose eine chirurgische Lungenbiopsie nötig, sofern das HRCT kein typisches UIP-Muster aufwies.[1]
Histologische Proben für die IPF-Diagnose müssen an mindestens drei Stellen entnommen werden und sie müssen groß genug sein, so dass der Pathologe die zu Grunde liegende Lungenarchitektur beurteilen kann. Kleine Biopsien, wie sie zum Beispiel während einer transbronchialen Lungenbiopsie (im Rahmen einer Bronchoskopie) durchgeführt werden, sind meistens nicht ausreichend für diesen Zweck. Aus diesem Grund sind meistens größere Biopsien notwendig, die chirurgisch im Rahmen einer Thorakotomie oder einer Thorakoskopie entnommen werden.[1][2]
Lungengewebe von Personen mit IPF weist zumeist ein charakteristisches histopathologisches UIP-Muster auf und ist somit das pathologische Gegenstück zur IPF.[1][4] Obwohl eine histopathologische UIP-Diagnose oft mit einer klinischen IPF-Diagnose einhergeht, können auch andere Krankheiten ein histologisches UIP-Muster sowie Fibrosen bekannter Herkunft aufweisen (z. B. Rheumakrankheiten).[1][2] Es gibt vier Schlüsselmerkmale für die UIP: interstitielle Patchwork-Fibrose, interstitielle Vernarbung, Honigwabenmuster und fibroblastische Foci.
Fibroblastische Foki sind dichte Ansammlungen von Myofibroblasten und Narbengewebe; zusammen mit dem Honigwabenmuster stellen sie die pathologischen Hauptbefunde dar, die eine UIP-Diagnose ermöglichen.
Bronchoalveoläre Lavage
[Bearbeiten | Quelltext bearbeiten]Die Bronchoalveoläre Lavage (BAL) ist ein gut zu tolerierendes diagnostisches Verfahren bei ILD.[29] Die Durchführung BAL-zytologischer Analysen (differentielle Zellzählungen) sollte bei der Abklärung von Patienten mit IPF im Ermessen des behandelnden Arztes liegen und je nach Einrichtungs-abhängiger Verfügbarkeit und Erfahrung durchgeführt werden. Die BAL kann alternative spezifische Diagnosen aufzeigen: Malignome, Infektionen, eosinophile Lungenentzündung, Langerhans-Zell-Histiozytose oder alveoläre Proteinose. Bei der Abklärung von Patienten mit Verdacht auf IPF trägt die BAL maßgeblich zum Ausschluss anderer Diagnosen bei.[5] Eine auffallende Lymphozytose (>30 %) schließt eine IPF-Diagnose vermutlich aus.[32]
Lungenfunktionstests
[Bearbeiten | Quelltext bearbeiten]Die Spirometrie zeigt eine Verminderung der Vitalkapazität (VC) mit entweder proportionaler Reduzierung der Luftvolumina oder Zunahme der Luftvolumina für die jeweils beobachtete Vitalkapazität auf. Letzterer Befund spiegelt eine zunehmende Lungenversteifung (verminderte Nachgiebigkeit) wider, die mit der Lungenfibrose einhergeht und zu einer erhöhten Lungenretraktion führt.[33]
Messungen statischer Lungenvolumina durch Bodyplethysmographie oder andere Methoden zeigen zumeist verringerte Lungenvolumina (restrictive Ventilationsstörung) auf. Dies spiegelt die Schwierigkeiten wider, mit denen man bei der Ausdehnung fibrotischer Lungen konfrontiert wird.
Die Diffusionskapazität der Lunge für Kohlenmonoxid (englisch Diffusing Capacity for Carbon Monoxide oder DLCO) ist bei IPF ausnahmslos eingeschränkt und kann in einem frühen oder leichten Krankheitsstadium die einzige Auffälligkeit darstellen. Die Einschränkung der DLCO ist dadurch bedingt, dass IPF-Patienten bei Belastung zu einer Sauerstoff-Entsättigung tendieren, was auch durch den 6-Minuten-Gehtest (englisch 6-minute walk test oder 6MWT) festgestellt werden kann.[1]
Begriffe wie „leicht“, „mittelschwer“ und „schwer“ werden gelegentlich benutzt, um die Krankheit in Stadien einzuteilen. Sie basieren für gewöhnlich auf Lungenfunktionstest-Messungen bei Ruheatmung.[1] Bei IPF-Patienten besteht jedoch weder in Bezug auf die Stadieneinteilung noch im Hinblick auf Kriterien und Werte, die dafür angewendet werden sollten, ein eindeutiger Konsens. Eine leichte bis mittelschwere IPF wird durch folgende funktionale Kriterien orientierend charakterisiert:[34][35][36][37]
- Forcierte Vitalkapazität (FVC): ≥50 %
- DLCO: ≥30 %
- 6MWT Entfernung ≥150 Meter.
Genetische Beratung bei familiärer IPF
[Bearbeiten | Quelltext bearbeiten]Geschätzte 10–15 % der IPF-Patienten leiden an einer Form der Krankheit, die in der Familie liegt und die daher familiäre Lungenfibrose genannt wird. Studien haben Genmutationen erkannt, die mit familiärer Lungenfibrose in Zusammenhang gebracht werden (s. o.). In ausgewählten IPF-Referenzzentren wird ein Test auf diese genetischen Mutationen angeboten.
Bei einer genetischen Beratung erhalten Patienten und ihre Familien Informationen zur Art, Vererblichkeit und zu den Folgen genetischer Erkrankungen. Diese Informationen können dazu verwendet werden, medizinische und persönliche Entscheidungen zu treffen und das Risiko für das Auftreten einer Erbkrankheit zu berechnen. In den Fällen, in denen mehr als ein Familienmitglied von Lungenfibrose betroffen ist, sollten eine genetische Beratung und ein Test zur Feststellung bekannter Mutationen durchgeführt werden. Eine genetische Beratung nach einem solchen Mutationstest ermöglicht eine individuelle Auslegung der Ergebnisse, d. h., was die Ergebnisse für die Gesundheit des Patienten bedeuten und wie sich dies auf andere Familienmitglieder auswirkt.
Prognose
[Bearbeiten | Quelltext bearbeiten]Der klinische Verlauf der IPF kann unvorhersehbar sein.[1][38][39] Die fortschreitende IPF geht mit einer geschätzten mittleren Überlebenszeit von 2 bis 5 Jahren nach Diagnosestellung einher.[1][2]
Die 5-Jahres-Überlebensrate bei IPF liegt bei 20–40 %;[39] die Mortalitätsrate ist somit höher als bei einer Anzahl von Malignomen, einschließlich Dickdarmkrebs, multiplem Myelom und Blasenkarzinom.[38][39]
Mögliche Verlaufsformen und therapeutische Überlegungen:[40]
Bei der Mehrzahl der Patienten kommt es zu einer langsamen, aber beständigen Verschlechterung ihres Zustandes („langsame Progression“). Manche Patienten zeigen einen langsamen Abfall („stabil“), während andere eine rasche Verschlechterung haben („rasche Progression“). Bei einer Minderheit der Patienten kann es zu einer unvorhersehbaren akuten Verschlechterung (akute Exazerbation) kommen, entweder aufgrund einer sekundären Komplikation wie Pneumonie oder aus unbekannten Gründen. Dieses Ereignis kann tödlich verlaufen oder die Patienten in einem erheblich schlechteren Zustand zurücklassen. Die jeweilige Häufigkeit dieser verschiedenen Spontanverläufe ist nicht bekannt.[40]
Dieses Modell wurde für IPF und andere ILDs verwendet und hat in Bezug auf die Sterblichkeitsprognose bei allen hauptsächlichen ILD-Subtypen gute Ergebnisse erzielt. Ein modifizierter ILD-GAP-Index wurde zur Anwendung auf ILD-Subtypen zur Berechnung erkrankungsspezifischer Überlebensraten entwickelt.[41]
Bei IPF-Patienten ist die Gesamtsterblichkeitsrate nach 5 Jahren hoch, die jährliche Rate bei Patienten mit milden bis mäßigen Lungenerkrankungen ist jedoch relativ gering. Deshalb wird in einjährigen klinischen Studien zur IPF-Behandlung normalerweise die Veränderung der Lungenfunktion (FVC) anstatt der Überlebensrate gemessen.[42]
Neben klinischen und physiologischen Parametern zur Vorhersage des Fortschreitens von IPF werden auch genetische und molekulare Eigenschaften mit IPF-Mortalität in Verbindung gebracht. So konnte z. B. bei IPF-Patienten mit einem spezifischen Genotyp im Mucin MUC5B-Genpolymorhismus (s. o.) ein langsamerer Rückgang der FVC und eine bedeutend höhere Überlebensrate nachgewiesen werden.[43][44]
Auch wenn diese Daten vom wissenschaftlichen Standpunkt aus betrachtet interessant sind, ist die Anwendung eines Prognosemodells auf der Grundlage spezifischer Genotypen in der klinischen Praxis noch nicht möglich.
Behandlung
[Bearbeiten | Quelltext bearbeiten]Behandlungsziele bei IPF sind im Wesentlichen Verbesserung der Symptomatik, Einhalt der Progression, Prävention akuter Exazerbationen und Verlängerung der Überlebensdauer. Präventive Maßnahmen (z. B. Impfungen) und Symptom-orientierte Behandlung sollten bei jedem Patienten möglichst früh eingeleitet werden.[45]
Pharmakologische Behandlung
[Bearbeiten | Quelltext bearbeiten]In der Vergangenheit wurde eine Vielzahl von Behandlungsmöglichkeiten für IPF untersucht, einschließlich γ-Interferon,[46] Endothelin-Rezeptoren-Hemmer (Bosentan,[47] Ambrisentan[48]) und Antikoagulanzien.[49] Da viele der früheren Studien auf der Annahme basierten, dass es sich bei der IPF um eine entzündliche Erkrankung handelt, werden diese Therapien nicht mehr als effektive Behandlungsmöglichkeiten in Betracht gezogen.
Pirfenidon
Pirfenidon ist ein kleines Molekül, das in experimentellen Fibrose-Modellen antioxidative, entzündungshemmende und anti-fibrotische Wirkung gezeigt hat.[50] Pirfenidon, das unter dem Namen Esbriet[51] vermarktet wird, ist in Europa für die Behandlung von Patienten mit leichter bis mittelschwerer IPF zugelassen. Es ist ebenfalls in Japan (Handelsname Pirespa), Südkorea, Indien, China, Kanada, Argentinien und Mexiko zugelassen.
Pirfenidon wurde in der Europäischen Union aufgrund der Ergebnisse von drei Phase III, randomisierten, doppelblinden, Placebo-kontrollierten Studien zugelassen, von denen eine in Japan und die anderen zwei in Europa und den USA (CAPACITY-Studien, PMID 21571362) durchgeführt wurden.[34][52]
Eine Durchsicht der Cochrane Library zeigte anhand von vier Studien, die Pirfenidon gegen ein Placebo in 1155 Patienten untersuchten, dass Pirfenidon zu einer signifikanten 30%igen Risikoreduktion der Krankheitsprogression führte.[53] Der Abfall von FVC oder VC wurde durch Pirfenidon ebenfalls signifikant verbessert, wenngleich eine geringe Verlangsamung der FVC-Abnahme nur in einer der beiden CAPACITY-Studien nachgewiesen werden konnte.[34] Aufgrund dieser nicht einheitlichen Ergebnisse verlangte die Arzneimittelzulassungsbehörde der USA, die Food and Drug Administration (FDA), eine dritte klinische Phase-III-Studie, ASCEND (NCT01366209, PMID 24836312).
Diese 2014 abgeschlossene Studie, die im New England Journal of Medicine veröffentlicht wurde, zeigte, dass Pirfenidon den Rückgang der Lungenfunktion und das Fortschreiten von IPF bedeutend reduzierte. [29] Die Daten der ASCEND-Studie wurden ebenfalls in einer präspezifizierten Analyse mit Daten aus den beiden CAPACITY-Studien kombiniert, woraus sich ergab, dass Pirfenidon das Sterberisiko im Laufe eines Behandlungsjahres um fast 50 % reduzierte.[36]
Anhand dieser Ergebnisse erhielt Pirfenidon die Bezeichnung als „Breakthrough Therapy“ (bahnbrechende Therapie) der US-amerikanischen Arzneimittelbehörde FDA. Diese Bezeichnung ist Medikamenten vorbehalten, die zur Behandlung schwerer oder lebensbedrohlicher Krankheiten dienen. Vorläufige klinische Nachweise zeigen, dass das Medikament an einem oder mehreren klinisch signifikanten Endpunkten eine bedeutende Verbesserung im Vergleich zu bereits bestehenden Therapien darstellt.[54]
Das Unternehmen, das Pirfenidon entwickelt hat, InterMune Inc., verwendete das Medikament im Zeitraum bis zu seiner Zulassung aus humanitären Gründen im Rahmen eines multizentrischen Härtefallprogramms (engl. EAP) in den Vereinigten Staaten.[55]
N-Acetylcystein und Triple-Therapie
Acetylcystein (NAC) ist ein Vorläufer von Glutathion, ein Antioxidans. Es besteht die Annahme, dass eine Behandlung mit NAC in hoher Dosierung ein Oxidantien-Antioxidantien Ungleichgewicht, das im Lungengewebe von IPF-Patienten auftritt, ausgleichen kann. In der ersten klinischen Studie mit 180 Patienten (IFIGENIA), konnte nachgewiesen werden, dass NAC über einen Beobachtungszeitraum von 12 Monaten die Abnahme der VC und DLCO vermindert, wenn es in Kombination mit Prednison und Azathioprin angewandt wird.[56]
Kürzlich wurde eine große randomisierte, kontrollierte Studie (PANTHER-IPF) von den National Institutes of Health (NIH) in den USA durchgeführt, um Dreifach-Therapie und NAC-Monotherapie bei IPF-Patienten zu untersuchen. Diese Studie ergab, dass die Kombination von Prednison, Azathioprin und NAC zu einem erhöhten Hospitalisierungs- und Sterberisiko führt. Das NIH erklärte daraufhin 2012, dass die Untersuchung der Dreifach-Therapie in der PANTHER IPF Studie frühzeitig abgebrochen wurde.[57]
Die Studie kam zu dem Ergebnis, dass „Acetylcystein im Vergleich zum Placebo keine bedeutenden Vorteile in Bezug auf die Aufrechterhaltung der FVC bei Patienten mit idiopathischer Lungenfibrose mit leichtem bis mäßigem Rückgang der Lungenfunktion“ bot.[58]
Diese Studie beurteilte zudem NAC allein und das Ergebnis für diesen Studienarm wurde vor Kurzem im New England Journal of Medicine veröffentlicht. Es zeigte sich jedoch, dass eine Monotherapie mit NAC ebenfalls keine bedeutenden vorteilhaften Auswirkungen auf Patienten mit leichter bis mittlerer IPF hat.[59]
Nintedanib
Nintedanib (Entwicklungscode BIBF 1120) hat eine Phase-II-Studie (TOMORROW)[60] und zwei Phase-III-Studien (INPULSIS-1 und INPULSIS-2) durchlaufen.[37] Nintedanib ist ein dreifacher Angiokinase-Inhibitor zur oralen Verabreichung, der die an der Regelung der Angiogenese beteiligten Rezeptor-Tyrosinkinasen hemmt: Fibroblasten-Wachstumsfaktorrezeptoren (FGFR), thrombozytäre Wachstumsfaktorrezeptoren (PDGFR) und vaskuläre endotheliale Wachstumsfaktorrezeptoren (VEGFR) ),[61] die auch an der Pathogenese von Fibrose und IPF beteiligt sind. In beiden Phase-III-Studien reduzierte Nintedanib signifikant den Rückgang der Lungenfunktion um etwa 50 % über ein Jahr hinweg. In Bezug auf die sekundären Endpunkte ergab sich ein bedeutender Anstieg des Zeitraums (verspätetes Einsetzen) bis zur ersten akuten Exazerbation (s. o.) in der mit Nintedanib behandelten Gruppe im Vergleich zur Placebo-Gruppe ausschließlich in der INPULSIS-2-Studie. In der INPULSIS-1-Studie wurde dieser Anstieg nicht beobachtet. Wie für Pirfenidon wurde das Gesuch auf Zulassung von Nintedanib von der FDA angenommen, und ein beschleunigtes Zulassungsverfahren („Priority Review“) wurde gewährt.[62] Nintedanib ist seit 2015 in den USA[63] und in Europa für alle Krankheitsstadien der IPF zugelassen.[64]
Zukünftige Therapieansätze
Eine Reihe an Wirkstoffen wird derzeit in klinischen Phase-II-Studien für IPF untersucht, darunter die monoklonalen Antikörper Simtuzumab, Tralokimunab und Lebrikizumab sowie FG-3019, ein Lysophosphatidsäure-Rezeptorantagonist (BMS-986020). Eine Phase-II-Studie zu STX-100 läuft ebenfalls.[65] Diese Moleküle dienen der Bekämpfung verschiedener Wachstumsfaktoren und Zytokine, die erwiesenermaßen eine Rolle in der Verbreitung, Aktivierung, Differenzierung oder im unangemessenen Überleben von Fibroblasten spielen.
Nicht-pharmakologische Behandlungen
[Bearbeiten | Quelltext bearbeiten]Lungentransplantation
Lungentransplantationen können für Patienten in Frage kommen, die aufgrund ihrer körperlichen Verfassung in der Lage sind, sich einer so großen Operation zu unterziehen. Bei IPF-Patienten, bei denen eine Lungentransplantation durchgeführt wurde, konnte das Mortalitätsrisiko im Vergleich zu Patienten, die sich noch auf der Warteliste befinden, um 75 % verringert werden.[66] Seit der Einführung des Lung Allocation Score (LAS), der Transplantations-Kandidaten anhand ihrer Überlebenswahrscheinlichkeit priorisiert, ist IPF in den USA zur häufigsten Indikation für eine Lungentransplantation avanciert.[67]
Symptomatische Patienten mit IPF, die jünger als 65 Jahre sind und einen Body-Mass-Index (BMI) von ≤26 kg/m² haben, sollten für eine Lungentransplantation vorgesehen werden. Es gibt jedoch keine genauen Daten, die zur Bestimmung des besten Zeitpunkts für eine Transplantation genutzt werden können. Obwohl es noch kontrovers ist, deuten die neuesten Daten an, dass eine bilaterale Lungentransplantation für IPF-Patienten geeigneter ist als die Transplantation eines einzelnen Lungenflügels.[68] Die 5-Jahres-Überlebensrate nach einer Lungentransplantation bei IPF wird auf 50 bis 56 % geschätzt.[1][69][70] Die Erfahrungen zur langfristigen Prognose der Lungentransplantation bei IPF im Vergleich zu anderen Indikationen sind uneinheitlich.[40]
Langzeit-Sauerstofftherapie (LTOT)
In den IPF-Richtlinien von 2011 ist die Sauerstofftherapie oder ergänzende Sauerstofftherapie für den Hausgebrauch eine wichtige Empfehlung für Patienten, die an klinisch signifikanter Ruhe-Hypoxämie leiden. Obwohl eine Verlängerung der Lebenserwartung durch Sauerstofftherapie nicht nachweisbar ist, so zeigen doch einige Daten, dass es zu einer Verbesserung der Belastungsfähigkeit kommt.[1][71]
Pulmonale Rehabilitation
Müdigkeit und Verlust an Muskelmasse sind häufige und beeinträchtigende Probleme für Patienten mit IPF. Pulmonale Rehabilitation kann die sichtbaren Symptome der IPF durch Stabilisierung oder Rückbildung extrapulmonaler Merkmale der Erkrankung lindern und den Funktionsstatus verbessern.[67][72]
Zur Rolle der pulmonalen Rehabilitation bei idiopathischer Lungenfibrose wurden bisher nur wenige Studien veröffentlicht, die meisten dieser Studien haben jedoch bedeutende kurzfristige Verbesserungen in Bezug auf die funktionelle Belastungstoleranz, Lebensqualität und Belastungsdyspnoe festgestellt.[73]
Typische Rehabilitationsprogramme beinhalten körperliches Training, Umstellung der Ernährung, Ergotherapie, Information und psychosoziale Beratung.
In den späten Stadien der Krankheit neigen IPF-Patienten dazu, körperliche Aktivitäten aufgrund fortschreitender Dyspnoe einzustellen. Wenn möglich, sollte dem Patienten davon abgeraten werden.
Palliative Behandlung
[Bearbeiten | Quelltext bearbeiten]Die Palliativbehandlung konzentriert sich in erster Linie auf die Linderung der Symptome und die Verbesserung der Lebensqualität des Patienten und nicht auf die Behandlung der Krankheit. Dies beinhaltet die anhaltende Therapie einer sich verschlechternden Symptomatik mit Opioiden bei akuter Luftnot und Husten. Ferner kann der palliative Einsatz von Sauerstofftherapie bei Dyspnoe in hypoxämischen Patienten hilfreich sein.
Palliativbehandlung umfasst ebenfalls die Linderung körperlicher und emotionaler Leiden sowie psychologische Unterstützung für Patienten und pflegende Angehörige.[1] Eine solche Versorgung kann nur individualisiert erfolgen und sollte als Ergänzung zur krankheitsbezogenen Therapie verstanden werden.[40]
Mit Fortschreiten der Krankheit können die Patienten unter Angst, psychischem Stress und Depressionen leiden. Psychologische Unterstützung sollte deshalb in Betracht gezogen werden. In einer aktuellen Studie mit ambulanten IDL-Patienten, einschließlich IPF-Patienten, haben der Grad der Depression, der funktionelle Status (ermittelt durch den Gehtest) als auch die Lungenfunktion gemeinsam zur Schwere der Dyspnoe beigetragen.[74]
In ausgesuchten Fällen mit besonders schwerer Dyspnoe kann die Verabreichung von Morphium in Betracht gezogen werden. Es kann die Dyspnoe lindern, Angstgefühle und Husten verringern, ohne die Sauerstoffsättigung signifikant zu senken.[75]
Management und Folgebehandlungen
[Bearbeiten | Quelltext bearbeiten]IPF wird oft fehldiagnostiziert, zumindest bis physiologische Befunde und/oder bildgebende Verfahren auf eine ILD schließen lassen, wodurch der Zugang zu geeigneter Behandlung verzögert wird.[67] Wenn man bedenkt, dass die IPF eine Krankheit mit einer mittleren Überlebenszeit von drei Jahren nach Diagnosestellung ist, sollte eine frühe Überweisung in eine Einrichtung, die über spezifisches Fachwissen verfügt, bei jedem Patienten mit Verdacht auf IDL oder mit bereits diagnostizierter IDL in Erwägung gezogen werden. Aufgrund der komplexen Differentialdiagnostik ist eine multidisziplinäre Diskussion zwischen Pulmologen, Radiologen und Pathologen, die Erfahrung in der Diagnose von ILD haben, für eine akkurate Diagnosestellung von äußerster Bedeutung.[1]
Nach IPF-Diagnosestellung und Wahl einer angemessenen, auf Symptome und Stadium der Krankheit ausgerichteten Therapie, sollte eine umfassende Verlaufsbeobachtung eingeleitet werden. Aufgrund des unvorhersehbaren Verlaufs der Krankheit und der hohen Komplikationsrate wie zum Beispiel Lungenkrebs (eine Rate von bis zu 25 % wurde unter IPF-Patienten gemeldet) ist eine Routineuntersuchung alle 3 bis 6 Monate notwendig, die Spirometrie (Bodyplethysmographie), Diffusionskapazitätstest, Röntgen-Thorax, 6MWT, Bewertung der Dyspnoe, Lebensqualität und Sauerstoffbedarf umfasst.
Aufgrund wachsender Erkenntnisse über Komplikationen und Begleiterscheinungen, die oft mit IPF in Verbindung stehen, ist eine regelmäßige Abklärung von Begleiterkrankungen notwendig. Die meisten spiegeln allerdings nur die üblichen Alterserkrankungen wider oder die Neben- und Wechselwirkungen der verwendeten Arzneimittel.
Akute Exazerbationen
[Bearbeiten | Quelltext bearbeiten]Akute Exazerbationen der IPF (AE-IPF) wurden bis 2016 definiert als: Ungeklärte Verschlechterung oder Entstehung von Atemnot innerhalb von 30 Tagen und neue radiologische Infiltrate im HRCT, die oft einen Hintergrund überlagern, der einem UIP-Muster entspricht. Eine internationale Arbeitsgruppe definierte im Jahr 2016 die Exazerbationen wie folgt: Akute, klinisch signifikante Verschlechterung der Atemfunktion, die durch den Nachweis einer neu aufgetretenen, ausgedehnten alveolären Anomalie charakterisiert wird.[76] Die überarbeiteten Diagnosekriterien sind:
- Frühere oder gleichzeitige Diagnose einer idiopathischen Lungenfibrose (wenn die Diagnose einer IPF nicht vorher gestellt wurde, kann dieses Diagnosekriterium bei der aktuellen Befundstellung durch den Nachweis von radiologischen oder histopathologischen Veränderungen, konsistent mit dem Befundmuster einer gewöhnlichen Lungenfibrose (UIP), erfüllt werden)
- Akute Verschlechterung der bestehenden Dyspnoe oder Entwicklung einer Dyspnoe typischerweise weniger als 1 Monat andauernd
- Computertomographie: neu aufgetretene beidseitige Milchglasverschattungen und/oder Konsolidierungen vor dem Hintergrund des Befundmusters einer „gewöhnlichen Lungenfibrose“ (Usual Interstitial Pneumonia, UIP). Wenn keine frühere Computertomographie zur Verfügung steht, kann die Einschränkung "neu" entfallen.
- Verschlechterung ist nicht vollständig durch Herzversagen oder Flüssigkeitsbelastung zu erklären.
Ereignisse, die aus klinischer Sicht die Definition der akuten Exazerbation der IPF erfüllen, jedoch nicht alle vier diagnostischen Kriterien aufgrund fehlender Computertomographie-Befunde erfüllen, sollten als „Verdacht auf akute Exazerbationen“ benannt werden.
Die jährliche Inzidenz von AE-IPF liegt bei allen Patienten zwischen 10 und 15 %. Die Prognose bei AE-IPF ist mit einer Mortalitätsrate von 78 % bis 96 % denkbar schlecht.[77] Andere Gründe für AE-IPF wie Lungenembolie, Herzinsuffizienz, Pneumothorax oder Infektionen müssen ausgeschlossen werden. Pulmonale Infektionen müssen durch endotracheale Aspirate oder BAL ausgeschlossen werden. Aufgrund des notfallmäßigen Zustandes der Patenten wird oft nur ein Teil der AE-IPF benötigten Untersuchungen durchgeführt. Von daher kann in diesen Fällen nur ein Verdacht auf AE gestellt werden. AE und vermutete AE sind klinisch identisch und in Hinsicht auf die Prognose und die Endpunkte der klinischen Studien als gleich wichtig zu betrachten.[78]
Viele Patienten, die eine akute Verschlechterung durchleben, benötigen eine Intensivbehandlung. Dies trifft insbesondere dann zu, wenn die respiratorische Insuffizienz mit hämodynamischer Instabilität, signifikanten Begleiterkrankungen oder schwerer Hypoxämie einhergeht. Die Sterblichkeit bei Klinikaufenthalten ist jedoch sehr hoch.[79] Mechanische Ventilation sollte erst nach genauer Abwägung der Langzeitprognose des Patienten und, wenn möglich, unter Berücksichtigung seiner eigenen Wünsche eingesetzt werden. Aktuelle Richtlinien raten jedoch von der Nutzung mechanischer Ventilation bei Patienten, die aufgrund der IPF an respiratorischer Insuffizienz leiden, ab.[1]
Initiativen für IPF Patienten: Die IPF-Charta
[Bearbeiten | Quelltext bearbeiten]Am 30. September 2014 wurde die IPF-Charta[80] im EU-Parlament vorgestellt. Die IPF-Charta ist eine Initiative der IPF-Patientenorganisationen und Ärzte aus verschiedenen Ländern. Mit dieser Europäischen Patienten-Charta fordern IPF Patientenorganisationen in ganz Europa politische Entscheidungsträger, Gesundheitsdienstleister, Geldgeber/Versicherer und nationale Regierungen auf, Maßnahmen zu ergreifen, die mehr Bewusstsein für die Idiopathische Lungenfibrose schaffen, die gleiche und verbesserte Behandlungsstandards gewährleisten und die einen gleichen Zugang sowie eine bessere Qualität in der Betreuung von IPF Patienten auf Europäischer Ebene schaffen. Mehr Informationen sind auf die Webseite zu finden.[81]
Bekannte Menschen mit idiopathischer Lungenfibrose
[Bearbeiten | Quelltext bearbeiten]- Marlon Brando, Film- und Theaterschauspieler
- Evel Knievel, weltbekannter Stuntman
- James Doohan, Schauspieler in Fernsehserie und Filmen der Star-Trek-Reihe
- Mette-Marit, Prinzessin von Norwegen[82]
- Brodie Lee, ehemaliger Wrestler
Idiopathische Lungenfibrose bei Tieren
[Bearbeiten | Quelltext bearbeiten]IPF tritt bei mehreren Hunde- und Katzenrassen gehäuft auf,[79] vor allem bei West Highland White Terriern.[83] Betroffene Tiere weisen viele klinische Merkmale wie menschliche Patienten auf, einschließlich zunehmende Belastungsintoleranz, erhöhte Atemfrequenz und letztendlich Atemnot.[84] Die Prognose ist im Allgemeinen schlecht.
Weblinks
[Bearbeiten | Quelltext bearbeiten]- Idiopathic Pulmonary Fibrosis (IPF) Community
- Pulmonary fibrosis foundation
- AIMIP Associazione Italiana Malattie Interstiziali o Rare del Polmone
- European IPF Registry (eurIPFreg) hat sich zu Europas führender Datenbank für Längsschnittdaten von IPF-Patienten, einschließlich Daten von Kontrollgruppen bestehend aus Patienten mit anderen Lungenkrankheiten
- Coalition for Pulmonary Fibrosis
- Die Aktivitäten der ILD CARE FOUNDATION konzentrieren sich auf die Vermehrung von Wissen, die Unterstützung von Forschung, der Förderung von Prävention und Beratung in Bezug auf interstetielle Lungenerkrankungen
- KnowIPFNow.com
- inIPF
- IPFtoday.com
- ipfcharter.org
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj G Raghu, HR Collard, JJ Egan et al.: An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. In: Am J Respir Crit Care Med., 183 (6), 2011, S. 788–824. doi:10.1164/rccm.2009-040GL. PMID 21471066
- ↑ a b c d e f g h i j k l m n o American Thoracic Society / European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. In: Am Respir Crit Care Med., 165 (2), January 2002, S. 277–304, PMID 11790668.
- ↑ a b EB Meltzer, PW Noble: Idiopathic pulmonary fibrosis. In: Orphanet J Rare Dis., 3 (1), 2008, S. 8. doi:10.1186/1750-1172-3-8. PMID 18366757. PMC 2330030 (freier Volltext).
- ↑ a b c d e Pulmonary Fibrosis Foundation. Prevalence and Incidence. Pulmonaryfibrosis.org; abgerufen am 11. April 2013.
- ↑ a b c d e f g G Raghu, D Weycker, J Edesberg, WZ Bradford, G Oster: Incidence and prevalence of idiopathic pulmonary fibrosis. In: Am. J Respir. Crit. Care Med, 174 (7), 2006, S. 810–816. doi:10.1164/rccm.200602-163oc.
- ↑ J Gribbin, RB Hubbard, I Le Jeune, CJ Smith, J West, LJ Tata: Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. In: Thorax 61 (11), 2006, S. 980–985. PMID 16844727. PMC 2121155 (freier Volltext).
- ↑ Eurostat News Release. (PDF; 177 kB) In: European demography, 110/2010, 27. Juli 2010.
- ↑ C Hyldgaard, O Hilberg, A Muller, E Bendstrup: A cohort study of interstitial lung diseases in central Denmark. In: Respir Med, 108 (5), 2014, S. 793–799. PMID 24636811.
- ↑ Jürgen Behr, Michael Kreuter, Marius M. Hoeper, Hubert Wirtz, Jens Klotsche, Dirk Koschel, David Pittrow: Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. In: European Respiratory Journal. 2. April 2015, ISSN 0903-1936, S. ERJ–02176–2014, doi:10.1183/09031936.00217614 (ersjournals.com [abgerufen am 13. Oktober 2016]).
- ↑ Jürgen Behr, Antje Prasse, Hubert Wirtz, Dirk Koschel, David Pittrow: Survival and course of lung function in the presence or absence of antifibrotic treatment in patients with idiopathic pulmonary fibrosis: long-term results of the INSIGHTS-IPF registry. In: The European Respiratory Journal. Band 56, Nr. 2, August 2020, ISSN 1399-3003, S. 1902279, doi:10.1183/13993003.02279-2019, PMID 32381492.
- ↑ Gabriela Leuschner, Jens Klotsche, Michael Kreuter, Antje Prasse, Hubert Wirtz: Idiopathic Pulmonary Fibrosis in Elderly Patients: Analysis of the INSIGHTS-IPF Observational Study. In: Frontiers in Medicine. Band 7, 2020, ISSN 2296-858X, S. 601279, doi:10.3389/fmed.2020.601279, PMID 33313046, PMC 7703706 (freier Volltext).
- ↑ Michael Kreuter, Jeff Swigris, David Pittrow, Silke Geier, Jens Klotsche, Hubert Wirtz, Andrea Prasse, Jürgen Behr: Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: INSIGHTS-IPF registry. In: Respiratory Research. Band 18, Nr. 1, 14. Juli 2017, S. 139, doi:10.1186/s12931-017-0621-y, PMID 28709421.
- ↑ INSIGHTS-ILD Register. 1. Dezember 2021, abgerufen am 23. Oktober 2023.
- ↑ Juergen Behr, Francesco Bonella, Andreas Günther, Dirk Koschel, Antje Prasse, David Pittrow, Jens Klotsche, Michael Kreuter, INSIGHTS-ILD Study Group: Investigating significant health trends in progressive fibrosing interstitial lung disease (INSIGHTS-ILD): rationale, aims and design of a nationwide prospective registry. In: BMC pulmonary medicine. Band 23, Nr. 1, 11. Februar 2023, ISSN 1471-2466, S. 64, doi:10.1186/s12890-023-02333-7, PMID 36774483, PMC 9922453 (freier Volltext).
- ↑ a b AL Olson, JJ Swigris: Idiopathic pulmonary fibrosis: diagnosis and epidemiology. In: Clinics in chest medicine, 33 (1), Mar 2012, S. 41–50. doi:10.1016/j.ccm.2011.12.001. PMID 22365244.
- ↑ KJ Williams: Gammaherpesviruses and Pulmonary Fibrosis: Evidence From Humans, Horses, and Rodents. In: Veterinary Pathology, 51 (2), March 2014, S. 372–384. doi:10.1177/0300985814521838. PMID 24569614.
- ↑ C García-Sancho, I Buendía-Roldán, MR Fernández-Plata, C Navarro, R Pérez-Padilla, MH Vargas, JE Loyd, M Selman, I Buendía-Roldán MR, Fernández-Plata et al.: Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. In: Respir Med., 105 (12), 2011, S. 1902–1990. PMID 21917441.
- ↑ a b S Harari, A Caminati: IPF: new insight on pathogenesis and treatment. In: Allergy, 65 (5), 2010, S. 537–553. doi:10.1111/j.1398-9995.2009.02305. PMID 20121758.
- ↑ a b H Loomis-King, KR Flaherty, BB Moore: Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. In: Curr Opin Pharmacol. 13 (3), 2013, S. 377–385. doi:10.1016/j.coph.2013.03.015. Epub 18. April 2013
- ↑ A Pardo, M Selman: Idiopathic pulmonary fibrosis: new insights in its pathogenesis. In: Int J Biochem Cell Biol., 34 (12), 2002, S. 1534–1538.
- ↑ M Selman, TE King, A Pardo: Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. In: Ann Intern Med. 134 (2), 2001, S. 136–151. PMID 11177318.
- ↑ a b Online Mendelian Inheritance in Man (OMIM) 178500 omim.org
- ↑ S Mathai et al.: Genetic susceptibility and pulmonary fibrosis. In: Curr Opin Pulm Med, 20 (5), 2014, S. 429–435. PMID 25022318.
- ↑ JA Kropski, DB Mitchell, C Markin et al.: A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. In: Chest, 6 Feb 2014. PMID 24504062.
- ↑ KR Flaherty, TE King, G Raghu, JP Lynch, TV Colby, WD Travis, BH Gross, EA Kazerooni et al.: Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? In: Am J Respir Crit Care Med., 170 (8), 2004, S. 904–910. doi:10.1164/rccm.200402-147OC. PMID 15256390.
- ↑ KR Flaherty, AC Andrei, TE King Jr, G Raghu, TV Colby, A Wells, N Bassily, K Brown et al.: Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? In: Am J Respir Crit Care Med., 175 (10), 2007, S. 1054–1060. PMID 17255566. PMC 1899268 (freier Volltext).
- ↑ diagnoseipf.com ( des vom 4. September 2014 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- ↑ a b KR Flaherty, JA Mumford, S Murray, EA Kazerooni, BH Gross, TV Colby, WD Travis, A Flint et al.: Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. In: Am J Respir Crit Care Med., 168 (5), 2003, S. 543–548. PMID 12773329.
- ↑ a b V Cottin, JF Cordier: Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis. In: Eur Respir J., 40 (3), 2012, S. 519–521. doi:10.1183/09031936.00001612, PMID 22941541.
- ↑ RP Baughman, RT Shipley, RG Loudon, EE Lower: Crackles in interstitial lung disease. Comparison of sarcoidosis and fibrosing alveolitis. In: Chest, 100 (1), 1991, S. 96–101. PMID 2060395.
- ↑ S. Ohshimo, F. Bonella u. a.: Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. In: American journal of respiratory and critical care medicine. Band 179, Nummer 11, Juni 2009, S. 1043–1047, doi:10.1164/rccm.200808-1313OC, PMID 19246718
- ↑ R Pellegrino, G Viegi, V Brusasco, RO Crapo, F Burgos, R Casaburi, A Coates, CP van der Grinten et al.: Interpretative strategies for lung function tests. In: Eur Respir J., 26 (5), 2005, S. 948–968. doi:10.1183/09031936.05.00035205. PMID 16264058.
- ↑ a b c PW Noble, C Albera, WZ Bradford, U Costabel, MK Glassberg, D Kardatzke, TE King, L Lancaster et al.: Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. In: The Lancet, 377 (9779), 2011, S. 1760–1769. doi:10.1016/S0140-6736(11)60405-4. PMID 21571362.
- ↑ FJ Martinez, S Safrin, D Weycker, KM Starko, WZ Bradford, TE King Jr, KR Flaherty, DA Schwartz et al. (IPF study group): The clinical course of patients with idiopathic pulmonary fibrosis. In: Ann Intern Med., 142 (12 Pt1), 2005, S. 963–967. PMID 15968010.
- ↑ a b TE King Jr., WZ Bradford, S Castro-Bernardini, EA Fagan, I Glaspole, MK Glassberg, E Gorina, PM Hopkins (for the ASCEND Study Group): A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. In: N Engl J Med., 370 (22), 2014, S. 2083–2092. PMID 24836312.
- ↑ a b L Richeldi, RM du Bois, G Raghu, A Azuma, KK Brown, M U Costabel, V Cottin, KR Flaherty (for the INPULSIS Trial Investigators): Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. In: N Engl J Med, 370 (22), 2014, S. 2071–2082. PMID 24836310.
- ↑ a b c JA Bjoraker, JH Ryu, MK Edwin, JL Myers, HD Tazelaar, DR Schroeder, KP Offord: Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. In: Am J Respir Crit Care Med., 157 (1), 1998, S. 199–203. doi:10.1164/ajrccm.157.1.9704130. PMID 9445300.
- ↑ a b c DS Kim, HR Collard, TE King: Classification and natural history of the idiopathic interstitial pneumonias. In: Proc Am Thorac Soc., 3 (4), 2006, S. 285–292. doi:10.1513/pats.200601-005TK. PMID 1673819. PMC 2658683 (freier Volltext).
- ↑ a b c d J Behr et al.: Leitlinie zur Diagnostik und Therapie der idiopathischen Lungenfibrose. In: Pneumologie, 67, 2013, S. 81–111.
- ↑ CJ Ryerson, E Vittinghoff, B Ley, JS Lee, JJ Mooney, KD Jones, BM Elicker, PJ Wolters et al.: Predicting Survival Across Chronic Interstitial Lung Disease: The ILD-GAP Model. In: Chest, 145 (4), 2014, S. 723–728. PMID 24114524.
- ↑ TE King Jr, C Albera, WZ Bradford, U Costabel, RM du Bois, JA Leff, SD Nathan, SA Sahn et al.: All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials. In: Am J Respir Crit Care Med, 189 (7), 2014, S. 825–831.
- ↑ AL Peljto, Y Zhang, TE Fingerlin, SF Ma, JG Garcia, TJ Richards, LJ Silveira, KO Lindell et al.: Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. In: JAMA, 309 (21), 2013, S. 2232–2239. PMID 23695349.
- ↑ CJ Stock, H Sato, C Fonseca, WA Banya, PL Molyneaux, H Adamali, AM Russell, CP Denton et al.: Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. In: Thorax, 68 (5), 2013, S. 436–441. PMID 23321605.
- ↑ JS Lee, S McLaughlin, HR Collard: Comprehensive care of the patient with idiopathic pulmonary fibrosis. In: Curr Opin Pulm Med., 17 (5), 2011, S. 348–354.
- ↑ TE King Jr, C Albera, WZ Bradford, U Costabel, P Hormel, L Lancaster, PW Noble, SA Sahn et al. (INSPIRE Study Group): Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis. In: Lancet, 374 (9685), 2009, S. 222–228. doi:10.1016/S0140-6736(09)60551-1. PMID 19570573.
- ↑ TE King Jr, KK Brown, G Raghu, RM du Bois, DA Lynch, F Martinez, D Valeyre, I Leconte et al.: BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. In: Am J Respir Crit Care Med., 184 (1), 2011, S. 92–99. doi:10.1164/rccm.201011-1874OC. PMID 21474646.
- ↑ G Raghu, J Behr, KK Brown, JJ Egan, SM Kawut, KR Flaherty, FJ Martinez, SD Nathan et al.: Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. In: Ann Intern Med., 158 (9), 2013, S. 641–649. doi:10.7326/0003-4819-158-9-201305070-00003. PMID 23648946.
- ↑ I Noth, KJ Anstrom, SB Calvert, J de Andrade, KR Flaherty, C Glazer, RJ Kaner, MA Olman: Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. In: Am J Respir Crit Care Med., 186 (1), 2012, S. 88–95. doi:10.1164/rccm.201202-0314OC. PMID 22561965.
- ↑ CJ Schaefer, DW Ruhrmund, L Pan, SD Seiwert, K Kossen: Antifibrotic activities of pirfenidone in animal models. In: Eur Respir Rev., 20 (120), 2011, S. 85–97.
- ↑ [1]
- ↑ H Taniguchi, M Ebina, Y Kondoh, T Ogura, A Azuma, M Suga, Y Taguchi, H Takahashi et al.: Pirfenidone in idiopathic pulmonary fibrosis. In: Eur Respir J., 35 (4), 2010, S. 821–829. doi:10.1183/09031936.00005209, PMID 19996196.
- ↑ P Spagnolo, C Del Giovane, F Luppi, S Cerri, S Balduzzi, EH Walters, R D’Amico, L Richeldi: Non-steroid agents for idiopathic pulmonary fibrosis. 2010. Cochrane Database Syst Rev. (9):CD003134. doi:10.1002/14651858.CD003134.pub2.
- ↑ InterMune Receives FDA Breakthrough Therapy Designation for Pirfenidone, an Investigational Treatment for IPF. Press release. Abgerufen am 8. April 2014: investor.intermune.com
- ↑ InterMune Announces Expanded Access Program for Pirfenidone to Treat Idiopathic Pulmonary Fibrosis (IPF) in the United States. Press release. Abgerufen am 8. April 2014:investor.intermune.com
- ↑ M Demedts, J Behr, R Buhl, U Costabel, R Dekhuijzen, HM Jansen, W MacNee, M Thomeer et al.: High-dose acetylcysteine in idiopathic pulmonary fibrosis. In: N Engl J Med. 353 (21), 2005, S. 2229–2242. doi:10.1056/NEJMoa042976. PMID 16306520.
- ↑ Commonly used three-drug regimen for idiopathic pulmonary fibrosis found harmful. Abgerufen am 11. April 2013
- ↑ J. Behr: Prednisone, azathioprine an N-acetylcysteine for pulmonary fibrosis. In: N Engl J Med 367 (9), 2012, S. 869–871. doi:10.1056/NEJMc1207471. PMID 22931324.
- ↑ The Idiopathic Pulmonary Fibrosis Clinical Research Network. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. In: N Engl J Med., 370 (22), 2014, S. 2093–2102. PMID 24836309.
- ↑ Luca Richeldi, Ulrich Costabel, Moises Selman, Dong Soon Kim, David M. Hansell: Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. In: The New England Journal of Medicine. Band 365, Nr. 12, 22. September 2011, ISSN 1533-4406, S. 1079–1087, doi:10.1056/NEJMoa1103690, PMID 21992121.
- ↑ BIBF 1120 Fact Sheet. ( des vom 3. März 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. (PDF; 110 kB) dl.groovygecko.net, abgerufen am 8. April 2014.
- ↑ Boehringer Ingelheim’s Investigational Therapy Nintedanib Receives FDA Breakthrough Therapy Designation. us.boehringer-ingelheim.com; Press release. Abgerufen am 8. April 2014.
- ↑ Pressemeldung der FDA zur Zulassung von Ofev in der Indikation IPF. Abgerufen am 13. Oktober 2016 (englisch).
- ↑ European Medicines Agency – Find medicine – Ofev. In: www.ema.europa.eu. Abgerufen am 13. Oktober 2016.
- ↑ coalitionforipf.org (Seite nicht mehr abrufbar, festgestellt im April 2018. Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.
- ↑ MJ Russo, A Iribarne, KN Hong, RR Davies, S Xydas, H Takayama, A Ibrahimiye, AC Gelijns, MD Bacchetta, F D’Ovidio, S Arcasoy, JR Sonett: High lung allocation score is associated with increased morbidity and mortality following transplantation. In: Chest. 137, Nr. 3, 2010, S. 651–657.
- ↑ a b c P Spagnolo, R Tonelli, E Cocconcelli, A Stefani, L Richeldi: Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges. In: Multidiscip Respir Med. 7, Nr. 1, 2012, S. 42.
- ↑ TJ George, GJ Arnaoutakis, AS Shah: Lung transplant in idiopathic pulmonary fibrosis. In: Arch Surg. 146, Nr. 10, 2011, S. 1204–1209.
- ↑ DP Mason, ME Brizzio, JM Alster, AM McNeill, SC Murthy, MM Budev, AC Mehta, OA Minai et al.: Lung transplantation for idiopathic pulmonary fibrosis. In: Ann Thorac Surg. 84, Nr. 4, 2007, S. 1121–1128. PMID 17888957.
- ↑ D Keating, B Levvey, T Kotsimbos, H Whitford, G Westall, T Williams, G Snell: Lung transplantation in pulmonary fibrosis challenging early outcomes counter balanced by surprisingly good outcomes beyond 15 years. In: Transplant Proc., 41 (1), 2009, S. 289–291. doi:10.1016/j.transproceed.2008.10.042. PMID 19249537.
- ↑ DA Morrison, JR Stovall: Increased exercise capacity in hypoxemic patients after long-term oxygen therapy. In: Chest. 102, Nr. 2, 1992, S. 542–550. PMID 1643945.
- ↑ JS Lee, S McLaughlin, HR Collard: Comprehensive care of the patient with idiopathic pulmonary fibrosis. In: Curr Opin Pulm Med. 17, Nr. 5, 2011, S. 348–354.
- ↑ K Kenn, R Gloeckl, J Behr: Pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis – a review. In: Respiration; international review of thoracic diseases. 86, Nr. 2, 2013, S. 89–99. doi:10.1159/000354112. PMID 23942353.
- ↑ CJ Ryerson, J Berkeley, VL Carrieri-Kohlman, SZ Pantilat, CS Landefeld, HR Collard: Depression and functional status are strongly associated with dyspnea in interstitial lung disease. In: Chest, 139 (3), 2011, S. 609–616.
- ↑ S Allen, S Raut, J Woollard, M Vassallo: Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis. In: Palliat Med., 19 (2), 2005, S. 128–130.
- ↑ Harold R. Collard, Christopher J. Ryerson, Tamera J. Corte, Gisli Jenkins, Yasuhiro Kondoh: Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. In: American Journal of Respiratory and Critical Care Medicine. Band 194, Nr. 3, 1. August 2016, ISSN 1535-4970, S. 265–275, doi:10.1164/rccm.201604-0801CI, PMID 27299520.
- ↑ R Agarwal, SK Jindal: Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review. In: Eur J Intern Med. 19, Nr. 4, 2008, S. 227–235.
- ↑ JB Stern, H Mal, O Groussard, O Brugière, A Marceau, G Jebrak, M Fournier: Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure. In: Chest. 120, Nr. 1, 2001, S. 213–219. doi:10.1378/chest.120.1.213.
- ↑ a b K Williams, D Malarkey, L Cohn, D Patrick, J Dye, G Toews: Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect. In: Chest. 125, Nr. 6, 2004, S. 2278–2288. doi:10.1378/chest.125.6.2278. PMID 15189952.
- ↑ "Launch of the first European IPF Patient Charter"
- ↑ ipfcharter.org ( des vom 22. Oktober 2014 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- ↑ Johanna Leufgens: Glitzernd wie eine Discokugel! Trotz unheilbarer Krankheit feiert sie durch die Nacht. Bunte, 15. November 2018, abgerufen am 16. November 2018.
- ↑ JA Webb, J Armstrong: Chronic idiopathic pulmonary fibrosis in a West Highland white terrier. In: Can Vet J. 43, Nr. 9, 2002, S. 703–705. PMID 12240528. PMC 339552 (freier Volltext).
- ↑ Canine Pulmonary Fibrosis. Akcchf.org. Abgerufen am 11. April 2013.