BLAST
| Programer(i) | Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, NCBI |
|---|---|
| Najnovija verzija | 2.5.0+ 12. септембар 2016. |
| Platforma | UNIX, Linux, Mac, Microsoft Windows |
| Tip | Bioinformatika |
| Licenca | Javno vlasništvo |
| Veb-sajt | blast |
U bioinformatici, BLAST (eng. Basic Local Alignment Search Tool) je algoritam za upoređivanje primarnih bioloških sekvenci, kao što su aminokiseline različitih proteina ili nukleotidi DNK sekvenci. BLAST pretraga omogućava istraživačima da porede niz sekvenci sa bibliotekama ili bazama podataka sekvenci, i identifikuju biblioteku sekvenci koja odgovara traženoj, sa određenom greškom.
Različite vrste BLAST-a su dostupne u zavisnosti od vrste sekvence koja se pretražuje. Na primer, nakon otkrića prethodno nepoznatog gena kod miševa, naučnik će obično primeniti BLAST pretragu ljudskih genoma da proveri da li ljudi sadrže slične gene; BLAST će prepoznati sekvence u ljudskom genomu koji su nalik mišjem zasnovanom na sličnosti sekvenci. BLAST algoritam i program su dizajnirani od strane Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, i David J. Lipman u Nacionalnom institutu za zdravlje i objavljen je u Journal of Molecular Biology 1990. i citiran više od 50,000 puta.[1]
Pozadina
[уреди | уреди извор]BLAST je jedan od najčešće koršćenih programa u bioinformatici za pretragu sekvenci.[2] On predstavlja fundamentalni problem u bioinformatici. Heuristički algoritam koji koristi je mnogo brži nego drugi pristupi, kao što je računanje optimalnog poravnanja. Naglasak na brzini je ključan aspekat algoritma, posebno na velikim trenutno dostupnim bazama genoma, iako noviji algoritmi mogu biti još brži.
Pre BLAST-a, FASTA je kreiran od strane David J. Lipman i William R. Pearson 1985.[3]
Pre brzih algoritama kao što su BLAST i FASTA, pretraga proteina i nukleinskih sekvenci je bila veoma vremenski zahtevna jer je korišćen potpun postupak poravnanja(npr., the Smith–Waterman algoritam).
Iako je BLAST brži od svih Smith-Waterman implementacija za većinu slučajeva, on ipak ne može da "garantuje optimalno poravnanje upita sa bazom sekvenci" kao Smith-Waterman algoritam. Optimalnost Smith-Waterman algoritma "obezbeđuje najveću tačnost i najpreciznije rezultate" po cenu vremena i resursa računara.
BLAST je vremenski efikasniji od FASTA jer pretražuje samo značajnije uzorake u sekvenci, ali sa određenom osetljivošću.
Primeri upotrebe BLAST-a:
- Koje vrste bakterija imaju protein koji je u vezi sa određenim proteinom koji ima poznatu aminokiselinu?
- Koji još geni predstavljaju proteine koji imaju strukturu sličnu onima koji su već otkriveni?
BLAST je često korišćen kao deo drugih algoritama koji zahtevaju približno poklapanje sekvenci.
Ulaz
[уреди | уреди извор]Ulaz predstavljaju sekvence (u FASTA formatu ili Genbank formatu) i težinska matrica.
Izlaz
[уреди | уреди извор]Izlaz BLAST algoritma može biti predstavljen na različite načine. Ovi formati mogu biti HTML, tekst i XML. Za NCBI web stranicu, podrazumevani format izlaza je HTML. Kada se izvrši BLAST algoritam na NCBI sajtu, rezultati su dati u grafičkom obliku i prikazuju pogotke, tabele prikazuju identifikatore sekvenci za pogotke zajedno sa pratećim podacima, kao i poravnanje sekvenci od interesa i pogotke dobijene korišćenjem odgovarajućeg BLAST vrednosnog sistema. Najjednostavnije za čitanje i najinformativnije su tabele.
Ako neko želi da pronađe sekvencu koje nema u bazama dostupnih javnosti putem izvora poput NCBI sajta, BLAST algoritam je moguće besplatno preuzeti sa interneta. Može se preuzeti sa NCBI sajta[4]. Dostupni su i komercijalni programi koji se mogu kupiti. Baze se mogu naći na NCBI sajtu, kao i na indeksu BLAST baza podataka[5].
Proces
[уреди | уреди извор]Korišćenjem heurističkih metoda, BLAST nalazi slične sekvence, lociranjem kratkih poklapanja između dve sekvence. Ovaj proces pronalaženja se naziva sejanje (eng. seeding). Nakon prvog poklapanja BLAST počinje da pravi lokalna poravnanja. Dok pokušava da nađe sličnost u sekvenci, skup čestih slova, poznat kao reč, je veoma važan. Na primer, pretpostavimo da sekvenca sadrži sledeći niz slova: GLKFA. Ako se BLAST pokrene pod normalnim uslovima, dužina reči bi bila 3 slova. U ovom slučaju, korišćenjem datog niza slova, dobijene reči bi bile GLK, LKF, KFA. BLAST-ov heuristički algoritam locira sve česte pojave troslovnih reči između zadate i pronađene sekvence. Ovaj rezultat se zatim koristi za pravljenje poravnanja. Nakon što je napravio reči za posmatranu sekvencu, ostale reči su takođe obrađene. Ove reči moraju da imaju zadovoljen prag T, u poređenju sa matricom vrednosti. Često korišćena matrica vrednosti za BLAST pretrage je BLOSUM62, iako optimalna matrica vrednosti zavisi od sličnosti sekvenci. Kada su reči, kao i okolne reči, procesuirane one se porede sa sekvencama iz baze u cilju pronalaska poklapanja. Prag T određuje da li konkretna reč ulazi u poravnanje. Kada je sejanje izvršeno, poravnanje koje je samo dužnine 3, je prošireno u oba smera pomoću BLAST algoritma. Svako proširenje utiče na rezultat poravnanja bilo povećanjem bilo smanjenjem. Ako je rezultat veći od unapred određenog T, poravnanje će biti uključeno u rezultat BLAST-a. Međutim, ako je rezultat manji od unapred određenog T, poravnanje će prestati da se širi, sprečavajući da se segmenti sa lošim poravnanjem uključe u rezultat BLAST-a. Primetimo da povećanjem T ograničavamo prostor koji možemo da pretražujemo, smanjujemo broj susednih reči, dok u isto vreme ubrzavamo BLAST.
Algoritam
[уреди | уреди извор]Za pokretanje programa, BLAST zahteva unos sekvence za pretragu, i sekvence sa kojom će upoređivati (takođe se naziva i “ciljana sekvenca”) ili baze koja sadrži više sekvenci. BLAST će naći više podsekvenci u bazi koje su slične podsekvenci upita. Obično, upitna sekvenca je dosta manja od baze, npr., upit može sadržati hiljadu nukleotida, dok baza sadrži nekoliko milijardi nukleotida.
Glavna ideja BLAST-a je da često postoji visoko rangirani segmentni parovi (High-scoring Segment Pairs (HSP)) sadržani u statistički bitnom poravnanju. BLAST traži visoko rangirana poravnanja sekvenci između upitne i posmatrane sekvence iz baze, koristeći heuristički pristup koji aproksimira Smith-Waterman algoritam. Međutim, iscrpni Smith-Waterman pristup je suviše spor za pretraživanje velikih baza genoma, kao što je GenBank. Stoga, BLAST algoritam koristi heuristički pristup koji je manje precizan od Smith-Waterman algoritma, ali preko 50 puta brži. Brzina i relativno dobra preciznost BLAST-a su među ključnim tehničkim inovacijama BLAST programa.
Pregled BLAST algoritma (protein-protein pretraga):[6] and CTGA2016
- Brisanje nekompleksnih regiona ili ponavljanje sekvenci iz upita.
- "Nekompleksni region" predstavlja region sekvenci sastavljen od nekoliko vrsti elemenata. Ovi regioni mogu da onemoguće program da nađe bitnu sekvencu u bazi, pa bi ih trebalo isfiltrirati. Ti regioni će biti obeleženi sa X(proteinske sekvence) ili N (nukleinske kiseline) i biće ignorisane od strane BLAST programa. Za filtriranje nekompleksnih regiona, za proteinske sekvence se koristi program SEG[7], a za DNK sekvence program DUST[8].
- Pravljenje k-slovne liste reči od upitne sekvence.
- Uzmimo k=3 kao primer. Izlistavamo reči dužine 3 iz upitne proteinske sekvence (k je obično 11 za DNK sekvencu) "sekvencijalno", dok sva slova upitne sekvence ne budu uključena. Metod je prikazan na slici 1.
Slika1 Metoda za pravljenje "k"-slovne liste upitnih reči
- Uzmimo k=3 kao primer. Izlistavamo reči dužine 3 iz upitne proteinske sekvence (k je obično 11 za DNK sekvencu) "sekvencijalno", dok sva slova upitne sekvence ne budu uključena. Metod je prikazan na slici 1.
- Lista mogućih reči koje se poklapaju.
- Ovaj korak je jedna od glavnih razlika između BLAST-a i FASTA. FASTA koristi sve česte reči nađene u koraku 2; međutim, BLAST koristi jedino visokovrednovane reči. Vrednosti su kreirane upoređivanjem reči dobijenih u koraku 2 sa svim preostalim rečima. Korišćenjem matrice vrednosti i poređenjem svih parova, postoji 20³ mogućih vrednosti za troslovne reči. Na primer, vrednost dobijena poređenjem PQG sa PEG i PQA je 15 i 12, redom. Za DNK reči, pogodak se računa kao +5, a promašaj kao -4, ili kao +2 i -3. Nakon toga, vrednost praga susednih reči T se koristi da smanji broj mogućih reči koje se poklapaju. Reči čije su vrednosti veće od praga T će ostati na listi tih reči, dok će reči čija je vrednost manja od ovog praga biti odbačene. Na primer, PEG se čuva, ali PQA je odbačen kada je T = 13.
- Organizacija visoko rangiranih reči u efikasno stablo pretrage.
- Ovo omogućava programu da brzo poredi visoko rangirane reči sa onima iz baze.
- Ponavljanje koraka 3 - 4 za svaku k-slovnu reč iz upitne sekvence.
- Pretraga baze radi pronalaženja egzaktnog poklapanja sa preostalim visoko rangiranim rečima.
- BLAST program pretražuje bazu po preostalim visoko rangiranim rečima, kao što je PEG, po svakoj poziciji. Ako se pronađe egzaktno poklapanje, ono se koristi za sejanje mogućih poravnanja bez odstupanja između upitne sekvence i sekvence iz baze.
- Proširivanje egzaktnog poklapanja u visoko rangirani segmentni par (HSP).
- Originalna verzija BLAST-a pravi dugačko poravnanje između upita i sekvenci iz baze u oba smera od pozicije gde je pronađeno egzaktno poklapanje. Proširenje ne prestaje sve dok nagomilana vrednost HSP-a ne počne da opada.
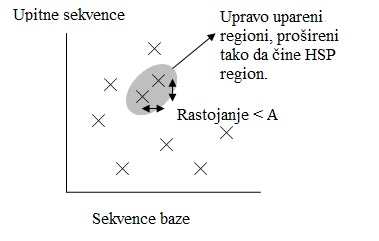
Slika2 Pozicija egzaktnih pogodaka - Radi uštede vremena, novija verzija BLAST-a, zvana BLAST2, je razvijena. BLAST2 prisvaja niže rangirane susedne reči da bi očuvao isti nivo osetljivosti za otkrivanje sličnih sekvenci. Stoga, lista mogućih poklapanja iz koraka 3 postaje duža. Dalje, regioni kod kojih je došlo do egzaktnog poklapanja, koji su na međusobnom rastojanju A na istoj dijagonali na slici 2, biće spojeni u jedan dugačak novi region. Konačno, novi regioni su prošireni na isti način kao u originalnoj verziji BLAST-a.
- Originalna verzija BLAST-a pravi dugačko poravnanje između upita i sekvenci iz baze u oba smera od pozicije gde je pronađeno egzaktno poklapanje. Proširenje ne prestaje sve dok nagomilana vrednost HSP-a ne počne da opada.
- Lista svih HSP u bazi čija je vrednost dovoljno velika da budu uzeti u obzir.
- Listamo HSP vrednosti koje su veće od empirijski utvrđenih granica vrednosti S. Proučavanjem distribucije vrednosti poravnanja dobijene poređenjem slučajnih sekvenci granična vrednost S može biti utvrđena tako da njena vrednost bude dovoljno velika da garantuje značajan ostatak HSP-a.
- Procena značajnosti HSP vrednosti.
- BLAST dalje pristupa statističkom značenju svake HSP vrednosti korišćenjem Gumbelove distribucije ekstremnih vrednosti (EVD)[9]. U poređenju sa Gumbel EVD, verovatnoća p za dostizanje S, koja je veća ili jednaka od x, data je sledećom jednačinom:
- gde je
- Statistički parametri i se procenjuju na osnovu vrednosti poravnanja bez rupa. Primetimo da i zavise od matrice zamene i kompozicije sekvence. i su efektivne dužine unosa i sekvence iz baze, redom. Dužina sekvence je skraćena na efektivnu dužinu da bi kompenzovala efekat ivice. Mogu se izračunati kao:
- gde je očekivana prosečna vrednost po poravnatom paru u poravnanju dve slučajne sekvence. Altschul i Gish dali su uobičajene vrednosti, , , i , za lokalno poravnanje bez rupa, korišćenjem BLOSUM62[10] kao matrice zamene. Korišćenje tipičnih vrednosti za dodeljivanje značajnosti se zove lookup-table metod, i on nije precizan. Očekivana vrednost E je broj koliko puta će nepovezana sekvenca iz baze dobiti vrednost S veću od x po verovatnoći. Očekivano E, koje je dobijeno u pretrazi baze od D sekvenci, dato je sa
- Šta više, kada , E može biti aproksimirano pomoću the Poisson distribucije kao
- Očekivana vrednost E dodeljuje značaj HSP vrednostima za lokalno poravnanje bez rupa, i to se predstavlja kao rezultat BLAST-a. Izračunavanja prikazana ovde se modifikuju ako se pojedinačni HSP kombinuju kao da predstavljaju rupičasto poravnanje, zbog varijacije statističkih parametara.
- BLAST dalje pristupa statističkom značenju svake HSP vrednosti korišćenjem Gumbelove distribucije ekstremnih vrednosti (EVD)[9]. U poređenju sa Gumbel EVD, verovatnoća p za dostizanje S, koja je veća ili jednaka od x, data je sledećom jednačinom:
- Spajanje dva ili više HSP regiona u duže poravnanje.
- Ponekad nađemo dva ili više HSP regiona u jednoj sekvenci iz baze, koji se mogu spojiti u duže poravnanje. Ovo obezbeđuje dodatnu evidenciju o relacijama između upita i sekvence iz baze. Postoje dva metoda: the Poisson metod i sum-of-scores metod, za poređenje značajnosti novodobijenih HSP regiona. Pretpostavimo da postoje dva kombinovana HSP regiona sa vrednostima (65, 40) i (52, 45), redom. The Poisson metod pridaje veći značaj skupu sa maksimalnom nižom vrednošću (45>40). Međutim, the sum-of-scores metod pridaje veći značaj prvom skupu, zato što je 65+40 (105) veće od 52+45(97). Originalni BLAST koristi the Poisson metod; BLAST2 i WU-BLAST kotiste the sum-of scores metod.
- Prikaz gapped Smith-Waterman lokalnog poravnanja u upitu i uparenih sekvenci iz baze.
- Originalni BLAST generiše samo cela poravnanja uključujući inicijalno uspostavljene HSP, čak i ako ima više od jednog HSP-a u sekvenci iz baze.
- BLAST2 koristi jednostruko rupičasto poravnanje koje uključuje sve inicijalno pronađene HSP regione.
- Prijava svakog pogotka čija je očekivana vrednost manja od parametra E.

Paralelni BLAST
[уреди | уреди извор]Verzija paralelnog BLAST-a koja koristi razdvojene baze je implementirana korišćenjem MPI i Pthreads, i prilagođena je različitim platformama, uključijući i Windows, Linux, Solaris, Mac OS X i AIX. Popularni pristup paralelizacije BLAST-a uključuje distribuirane upite, segmentaciju heš tabela, paralelno računanje i segmentaciju baza. Baze su podeljene na jednake delove i čuvaju se na lokalnim čvorovima. Svaki upit je pokrenut na svim čvorovima paralelno i izlazni fajlovi su spojeni u finalni izlaz.
Program
[уреди | уреди извор]BLAST program može biti, ili preuzet, ili pokrenut iz komandne linije, ili se može koristiti besplatno onlajn. BLAST-ov web server, održavan od strane NCBI, dozvoljava svakome sa web pretraživačem da izvršava slične pretrage na konstantno ažuriranoj bazi proteina i DNK, koja uključuje većinu organizama.
BLAST program je otvorenog koda, što daje svima mogućnost da ga koriste i menjaju. Ovo je dovelo do nastanka više varijanti BLAST programa.
Danas su dostupne različite korisne varijacije BLAST-a, koje mogu biti korišćene u zavisnosti od onoga šta želimo da uradimo i sa čime radimo. Ove varijacije programa su različite po pitanju upitnih sekvenci, baze koja se pretražuje i šta se upoređuje. Ovi programi i njihovi opisi su izlistani ispod:
BLAST je zapravo familija programa (sve su uključene u blastall izvršavanje). Ovo uključuje:[11]
- Nukleotid-nukleotid BLAST (blastn)
- Ovaj program, kome se zadaje DNK upit, vraća najsličniju DNK sekvencu iz baze koju je korisnik odabrao.
- Protein-protein BLAST (blastp)
- Ovaj program, kome se zadaje proteinski upit, vraća najsličniju proteinsku sekvencu iz baze koju je korisnik odabrao.
- Position-Specific Iterative BLAST (PSI-BLAST) (blastpgp)
- Ovaj program se koristi za pronalaženje daljih rođaka proteina. Prvo se kreira lista svih blisko povezanih proteina. Ovi proteini se kombinuju u opštu “profil" sekvencu, koja sumira značajne osobine prisutne u tim sekvencama. Upit na bazi proteina koristi taj profil, i pronalazi se veća grupa proteina. Ova veća grupa se koristi za pravljenje novog profila i proces se ponavlja.
- Uključivanjem povezanih proteina u pretragu, PSI-BLAST je osetljiviji u izboru rođaka iz filogenetskog stabla od običnog protein-protein BLAST-a.
- Nucleotide 6-frame translation-protein (blastx)
- Ovaj program poredi šest frejmova konceptualnom translacijom nukleotida upitne sekvence sa proteinom iz baze.
- Nucleotide 6-frame translation-nucleotide 6-frame translation (tblastx)
- Ovaj program je najsporiji iz BLAST famiije. On translira upitnu sekvencu nukleotida u svih šest mogućih frejmova i poredi ih sa šest frejmova translacijom nukleotida upitne sekvence sa proteinom iz baze. Svrha ovoga je da nađe veoma daleke veze između sekvenci nukleotida.
- Protein-nucleotide 6-frame translation (tblastn)
- Ovaj program poredi upit sa svih šest frejmova sa sekvencom nukleotida iz baze.
- Veliki broj upitnih sekvenci (megablast)
- Pri upoređivanju velikog broja ulaznih sekvenci putem BLAST-a komandne linije, "megablast" je mnogo brži od višestrukog pokretanja BLAST-a. On vrši konkatenaciju više ulaza kako bi formirao veliku sekvencu pre pretrage, a zatim se naknadnom analizom dobijaju konačni rezultati.
Alternativne verzije
[уреди | уреди извор]Verzija dizajnirana za upoređivanje velikih genoma ili DNK je BLASTZ.
CS-BLAST (ContSxt-Specific BLAST) je proširena verzija BLAST-a za pretragu proteinskih sekvenci koja pronalazi dvostruko više daleko povezanih sekvenci od BLAST-a za isto vreme i sa istom stopom greške. U CS-BLAST-a, verovatnoća mutacije između aminokiselina ne zavisi samo od jedne aminokiseline kao u BLAST-u, već i od konteksta lokalne sekvence. Vašington Univerzitet napravio je alternativnu verziju NCBI BLAST-a, zvanu WU-BLAST. Autorska prava pripadaju Advanced Biocomputing, LLC.
2009. NCBI je objavio novu seriju BLAST izvršnih programa, C++ zasnovani BLAST+,[12], i objavio je paralelnu verziju do 2.2.26. Počevši sa verzijom 2.2.27 (April 2013), samo BLAST+ izvršni programi su dostupni. Među izmenama je i zamena blastall komande za više različitih komandi za različite BLAST programe, i promene u rukovanju opcijama.
Alternative BLAST-a
[уреди | уреди извор]Ekstremno brza, ali znatno manje osetljiva, alternativa BLAST-u je BLAT (eng. Blast Like Alignment Tool). Dok BLAST vrši linearnu pretragu, BLAT se oslanja na k-mer indeksiranje baze, i na taj način često može brže da pronađe seme. Još jedan program sličan BLAT-u je PatternHunter.
Napretkom tehnologija sekvencioniranja kasnih 2000-tih, pronalaženje veoma sličnih nukleotida postaje važan problem. Novi programi poravnanja skrojeni za ovu specifičnu upotrebu koriste BWT-indeksiranje ciljane baze (obično genoma). Ulazna sekvenca može biti mapirana vrlo brzo, a izlaz je obično u vidu BAM fajla. Primeri programa poravnanja su BWA, SOAP i Bowtie.
Za identifikaciju proteina, traženje poznatih domena (npr. Pfam) povezivanjem sa Hidden Markov Models je popularna alternativa, kao što je npr. HMMER.
Alternativa BLAST-u za poređenje dve banke sekvenci je KLAST[13]. Rezultati KLAST-a su veoma slični rezultatiima BLAST-a, ali KLAST je značajno brži i sposobniji da poredi velike skupove sekvenci sa malim utroškom memorije.
Primena BLAST
[уреди | уреди извор]BLAST se može koristiti u više svrha. Ovo uključuje identifikaciju vrsti, lociranje domena, uspostavljanje filogenije, DNK mapiranje i poređenje.
- Identifikacija vrsti
- Korišćenjem BLAST-a, moguće je tačno identifikovati vrstu ili naći odgovarajuću vrstu. Ovo može biti korisno, na primer, za rad sa DNK sekvencama nepoznatih vrsti.
- Lociranje domena
- Proteinske sekvence je moguće proslediti kao ulaz BLAST-u da bi se locirali poznati domeni te sekvence.
- Uspostavljanje filogenije
- Korišćenjem rezultata BLAST-a moguće je kreirati filogenetsko stablo korišćenjem BLAST web-strane. Filogenija zasnovana samo na BLAST-u je manje pouzdana nego druge, za tu svrhu napravljene, filogenetske metode, tako da bi trebalo da bude korišćena samo kao prvi produkt analize.
- DNK mapiranje
- Pri radu sa poznatim vrstama i traženja sekvenci gena na nepoznatoj lokacijii, BLAST može porediti hromozomske pozicije hromozomske pozicije.
- Poređenje
- Pri radu sa genima, BLAST može da locira česte gene u dve povezane vrste, i može da se koristi za mapiranje razlika između organizama.
Poređenje BLAST-a i Smith-Waterman procesa
[уреди | уреди извор]Dok se i Smith-Waterman i BLAST koriste za pronalaženje odgovaraćujih sekvenci pretragom i poređenje upitne sekvence sa onim iz baza, oni imaju razlike.
Iako je BLAST zasnovan na heurističkom algoritmu, rezultati dobijeni upotrebom BLAST-a, u terminima broja pronađenih pogodaka, možda neće dati najbolje rezultate, jer neće pronaći sva podudaranja sa bazom.
Bolja alternativa za pronalaženje najboljeg mogućeg rešenja bila bi korišćenje Smith-Waterman algoritma. Ovaj metod se razlikuje od BLAST-a u dve oblasti, preciznosti i brzni. The Smith-Waterman obezbeđuje veću preciznost, jer pronalazi podudaranja koja BLAST ne može, jer ne preskače nijednu informaciju. Međutim, u poređenju sa BLAST-om, troši više vremena i zahteva veću količinu kompjuterskih resursa. Pronađene su tehnologije koje mogu znatno da ubrzaju Smith-Waterman proces. Te tehnologije uključuju FPGA čipove i SIMD technologiju.
Za dobijanje boljih rezultata BLAST-a, podrazumevana podešavanja se mogu promeniti. Ne postoji siguran način za menjanje podešavanja kako bi se obezbedio najbolji rezultat za datu sekvencu.
Vidi još
[уреди | уреди извор]Reference
[уреди | уреди извор]- ^ Altschul, Stephen; Gish, Warren; Miller, Webb; Myers, Eugene; Lipman, David (1990). „Basic local alignment search tool”. Journal of Molecular Biology. 215 (3): 403—410. PMID 2231712. doi:10.1016/S0022-2836(05)80360-2.
- ^ Casey, R. M. (2005). „BLAST Sequences Aid in Genomics and Proteomics”. Business Intelligence Network.
- ^ Lipman, DJ; Pearson, WR (1985). „Rapid and sensitive protein similarity searches”. Science. 227 (4693): 1435—41. PMID 2983426. doi:10.1126/science.2983426.
- ^ BLAST+ executables
- ^ Index of BLAST databases (FTP)
- ^ Mount, D. W. (2004). Bioinformatics: Sequence and Genome Analysis (2nd изд.). Cold Spring Harbor Press. ISBN 978-0-87969-712-9.
- ^ SEG
- ^ DUST
- ^ Gumbel extreme value distribution (EVD)
- ^ BLOSUM62
- ^ „Program Selection Tables of the Blast NCBI web site”.
- ^ Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T. L. (2009). „BLAST+: Architecture and applications”. BMC Bioinformatics. 10: 421. PMC 2803857
. PMID 20003500. doi:10.1186/1471-2105-10-421.
- ^ „KLAST”. Архивирано из оригинала 26. 05. 2017. г. Приступљено 9. 05. 2016.
Literatura
[уреди | уреди извор]- Mount, D. W. (2004). Bioinformatics: Sequence and Genome Analysis (2nd изд.). Cold Spring Harbor Press. ISBN 978-0-87969-712-9.
- Baxevanis, Andy (2005). „Chapter 11: Assessing Pairwise Sequence Similarity: BLAST and FASTA”. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins, Third Edition. New York: John Wiley & Sons.
- Wheeler, David; Bhagwat, Medha (2007). „Chapter 9: BLAST QuickStart”. Ур.: Bergman, Nicholas H. Comparative Genomics Volumes 1 and 2. Methods in Molecular Biology. 395-396. Totowa, NJ: Humana Press. PMID 21250292.
- Mount, DW (1. 7. 2007). „Using the Basic Local Alignment Search Tool (BLAST)”. Cold Spring Harbor Protocols. 2007 (14): pdb.top17. PMID 21357135. doi:10.1101/pdb.top17.
Spoljašnje veze
[уреди | уреди извор]- Званични веб-сајт
- BLAST+ executables — free source downloads
- Biological Sequence Analysis I: Andy Baxevanis' lecture from NHGRI's Current Topics in Genome Analysis series, covering contemporary areas in genomics and bioinformatics
- What's behind BLAST?: talk by Gene Myers (slides and video)
| Државне | |
|---|---|
| Остале | |