Sindrome di Miller
La sindrome di Miller, conosciuta anche come sindrome di Genée-Wiedemann, è una malattia genetica rara, dall'incidenza incerta, a trasmissione autosomica recessiva, che consiste in anomalie cranio-facciali, degli arti e degli occhi.
| Sindrome di Miller | |
|---|---|
 | |
| Malattia rara | |
| Specialità | genetica clinica |
| Classificazione e risorse esterne (EN) | |
| OMIM | 263750 e 263750 |
| MeSH | C537680 |
| Sinonimi | |
| Sindrome di Genée-Wiedemann Sindrome di Wilderwanck-Smith Sinostosi acrofacciale postassiale | |
Storia
modificaLa sindrome venne descritta la prima volta dai medici tedeschi Ekkert Genée e Hans-Rudolf Wiedemann, rispettivamente nel 1969 e nel 1975. Il primo riteneva che si trattasse della sindrome di Treacher Collins espressa a un livello fenotipico estremo (nominandola disostosi mandibolofacciale),[1] mentre Wiedemann la descrisse come una malattia a sè stante.[2] Ulteriori casi clinici vennero riportati da Wilderwanck, sempre nel 1975, e da Miller et al., nel 1979.[3]
Eziologia
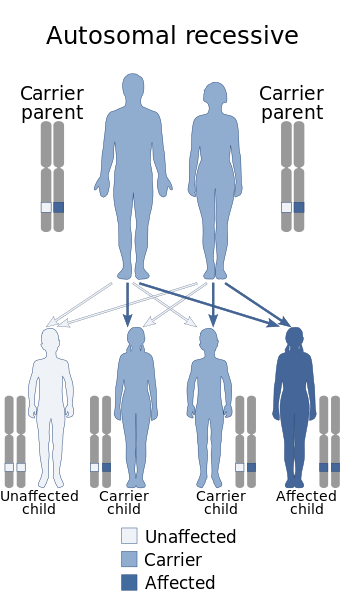
modificaLa sindrome è causata da una mutazione del gene DHODH,[4] deputato alla codifica dell'enzima diidroorotato deidrogenasi, che catalizza l'ossidazione ubichinone-mediata del diidroorotato in orotato, quarto step del processo di sintesi de novo delle pirimidine. Questo enzima è collocato nella porzione esterna della membrana mitocondriale interna. Il gene DHODH è situato sul braccio lungo del cromosoma 16, in corrispondenza del locus genico 16q22. La prima mutazione di questo gene documentata in biologia venne individuata da Morgan nel 1910 e riguarda il genoma della mosca Drosophila melanogaster; gli esemplari con questa mutazione genetica presentavano anomalie delle ali, delle zampe posteriori e un'oogenesi difettosa.[5] La mutazione che causa la sindrome di Miller negli esseri umani è trasmessa in modo autosomico recessivo.
Clinica
modificaSegni e sintomi
modificaI segni clinici comprendono una marcata micrognazia (conseguenza dello scarso sviluppo delle ossa della mandibola), cheiloschisi o palatoschisi, ipoplasia o aplasia delle porzioni terminali degli arti, coloboma della palpebra e politelia. Ulteriori manifestazioni della sindrome possono essere: rime palpebrali ribassate, ipoplasia malare, malformazioni alle orecchie e l'allargamento anomalo del dorso nasale.
La sindrome può manifestarsi anche con altre condizioni anomale, come la presenza di vertebre in sovrannumero e/o segmentate in modo anomalo, difetti a carico delle costole, difetti cardiaci (pervietà del dotto di Botallo, difetto interventricolare e difetti del setto interatriale). Le anomalie a carico della gabbia toracica possono portare a infezioni croniche, in genere polmoniti. Si può inoltre riscontrare la presenza di una sola arteria ombelicale, agenesia del diaframma, ipoplasia del femore, difetti nel processo di ossificazione dell'ischio e del pube, lingua bilobata, ipoplasia polmonare (conseguente allo scarso sviluppo delle ossa del torace) e problemi renali.
Diagnosi differenziale
modificaLa sindrome di Miller entra in diagnosi differenziale con la sindrome di Treacher Collins, con la sindrome di Nager (una forma di craniodisostosi pre-assiale) e con altre, rare forme di craniodisostosi su base genetica.
Note
modifica- ^ (FR) Ekkert Genée, Une forme de dysostose mandibulo-faciale, in Journal De Génétique Humaine, vol. 17, 1969, pp. 45–52.
- ^ (DE) Hans-Rudolf Wiedemann, Missbildungs-Retardierungs-Syndrom mit Fehlen des 5. Strahls an Händen und Füssen, Gaumenspalte, dysplastischen Ohren und Augenlidern und radioulnarer Synostose, in Klin. Padiatr., vol. 185, n. 3, 1975, pp. 181-186, PMID 4795571.
- ^ (EN) M. Miller, R. Fineman et al., Postaxial acrofacial dysostosis syndrome, in The Journal of Pediatrics, vol. 95, n. 6, dicembre 1979, pp. 970–975, DOI:10.1016/S0022-3476(79)80285-1, PMID 501501.
- ^ (EN) Ng SB, Buckingham KJ, Lee C, Bigham AW et al., Exome sequencing identifies the cause of a mendelian disorder, in Nature Genetics, vol. 42, n. 1, gennaio 2010, pp. 30-35, DOI:10.1038/ng.499, PMC 2847889, PMID 19915526.
- ^ (EN) T.H. Morgan, Sex limited inheritance in drosophila, in Science, vol. 32, n. 812, luglio 1910, pp. 120–122, DOI:10.1126/science.32.812.120, PMID 17759620.