Malattia di Wilson
La malattia di Wilson (un tempo detta anche morbo[1] di Wilson), o degenerazione epatolenticolare, è una malattia genetica, trasmessa in modo autosomico recessivo, che determina l'accumulo di rame nei tessuti; i sintomi si manifestano a livello neurologico-psichiatrico e soprattutto a livello del fegato. Nei bambini l'esordio è spesso con sintomi epatici, mentre negli adulti sono i sintomi neurologici a esordire per primi.[2] Si può manifestare tra i 5 e i 40 anni, e gli esordi precoci corrispondono a un decorso più grave, pericoloso e rapido.[3] I sintomi appaiono in genere tra i 6 e i 20 anni, anche se in alcuni casi sono stati descritti primi sintomi in pazienti molto più anziani.[4]
| Malattia di Wilson | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RC0150 |
| Specialità | endocrinologia |
| Eziologia | genetica |
| Classificazione e risorse esterne (EN) | |
| ICD-9-CM | 275.1 |
| OMIM | 277900 |
| MeSH | D006527 |
| MedlinePlus | 000785 |
| eMedicine | 183456 e 1153622 |
| GeneReviews | Panoramica |
| Sinonimi | |
| degenerazione epatolenticolare degenerazione lenticolare progressiva | |
| Eponimi | |
| Samuel Alexander Kinnier Wilson | |
La condizione è dovuta a una mutazione nella proteina della malattia di Wilson (gene ATP7B). Una singola copia anomala del gene è presente in una persona su cento, senza determinare alcun sintomo, essendo la patologia causata da un gene recessivo (si tratta di portatori sani della malattia). Se un individuo eredita il gene da entrambi i genitori, è a rischio di manifestare la patologia. La malattia di Wilson ha un'incidenza di 2,66/100 000 con una prevalenza di 6,21/100 000.[4] La malattia prende il nome da Samuel Alexander Kinnier Wilson (1878-1937), neurologo inglese che per primo descrisse la condizione nel 1912.[5]
Senza cura, la malattia può essere facilmente letale in pochi anni, tuttavia esistono farmaci efficaci in grado di controllarla; la cura prevede l'utilizzo di farmaci chelanti che riducono l'assorbimento di rame e ne rimuovono l'eccesso dall'organismo, farmaci di mantenimento, a volte fisioterapia, e un'adeguata dieta povera di rame, ma occasionalmente è necessario anche un trapianto di fegato in caso di grave insufficienza epatica.[6] A causa della grande variabilità dei sintomi e del decorso, la diagnosi è raramente tempestiva, per cui i pazienti aspettano anche anni, con peggioramento dei sintomi, prima di sapere di esserne affetti e potersi curare. Comunque, se trattata adeguatamente, la malattia regredisce in buona parte e non riduce l'aspettativa di vita dei pazienti, che rimane identica a quella della popolazione sana, così come si può mantenere una buona qualità di vita; tuttavia il trattamento farmacologico e il monitoraggio devono proseguire in maniera costante e cronica per tutta la durata della vita.[2]
Storia
modificaLa malattia porta il nome del medico britannico Samuel Alexander Kinnier Wilson (1878-1937), un neurologo che ha descritto la condizione, comprese le mutazioni patologiche nel cervello e nel fegato, nel 1912.[5] Il lavoro di Wilson era stato preceduto dalle relazioni del neurologo tedesco Carl Westphal che, nel 1883, definì la condizione come "pseudosclerosi", da quelle del neurologo britannico William Gowers (nel 1888), e dagli studi effettuati nel 1898 da Adolph Strümpell che notò la cirrosi epatica.[7] Nel 1958, il neuropatologo John Nathaniel Cumings, studiò la correlazione tra l'accumulo di rame sia nel fegato sia nel cervello.[8] La presenza di emolisi è stata notata nel 1967.[9]
Cumings insieme e contemporaneamente al neurologo neozelandese Derek Denny-Brown, che lavorava negli Stati Uniti, per primi identificarono, nel 1951, un trattamento efficace con il Dimercaprol.[10][11] Il farmaco veniva iniettato ed è stata una delle prime terapie disponibili in neurologia, una specialistica questa che storicamente era in grado di osservare e diagnosticare, ma che disponeva di pochi trattamenti efficaci.[7][12] Il primo farmaco efficace come agente chelante orale, la penicillamina, venne scoperta nel 1956 dal neurologo britannico John Walshe.[13] Nel 1982, Walshe introdusse anche la trientina[14] e fu il primo a sviluppare il tetratiomolibdato per uso clinico[15]. La terapia a base di acetato di zinco comparve inizialmente nei Paesi Bassi, dove Schouwink e Hoogenraad l'hanno utilizzata rispettivamente nel 1961 e nel 1970; questa terapia fu ulteriormente sviluppata successivamente da Brewer e colleghi dell'Università del Michigan.[16][17]
La base genetica della malattia di Wilson e il collegamento con le mutazioni del gene ATP7B è stata chiarita negli anni ottanta e anni novanta da diversi gruppi di ricerca; tra gli scopritori del gene anche due pediatri italiani, Enrico Parano e Lorenzo Pavone.[18][19]
Segni e sintomi
modificaIl rame si accumula principalmente nel fegato e nel cervello; a ciò conseguono le manifestazioni rispettivamente epatiche e neuropsichiatriche che rappresentano i principali segni che conducono alla diagnosi.[6] Pazienti con problemi di fegato son soliti rivolgersi prima all'analisi medica, generalmente in età infantile o adolescenziale, rispetto a quelli con sintomi neurologici o psichiatrici, che invece vi si rivolgono dai venti anni in su, quando il quadro clinico è già maggiormente compromesso. Alcuni soggetti affetti dalla malattia sono identificati solo perché imparentati con individui a cui già è stata diagnosticata la malattia di Wilson; successivamente al test, si scopre spesso che molti di questi pazienti hanno già sperimentato i sintomi della malattia senza però ricevere una diagnosi.[20]
Danni epatici
modificaI disturbi epatici possono presentarsi con stanchezza, emorragia, dolore addominale o confusione (a causa dell'encefalopatia epatica, cfr. i sintomi neuropsichiatrici della patologia) e ipertensione portale. Inoltre, nel caso in cui la pressione nella vena porta aumenti considerevolmente, appaiono le varici esofagee (vasi sanguigni che sanguinano nell'esofago), splenomegalia (aumento di volume della milza) e ascite (accumulo di fluidi nella cavità addominale). All'esame, possono essere rilevati segni di disagi cronici del fegato come la teleangectasia (vasi sanguigni distesi, di solito sul torace). Spesso nei pazienti che riportano i sintomi è diagnosticata una cirrosi epatica, con fibrosi. Sebbene la maggior parte delle persone con cirrosi abbiano un rischio superiore di sviluppare un tumore del fegato, tale rischio è relativamente basso nei pazienti affetti da malattia di Wilson.[6]
Il 5% circa dei pazienti ricevono una diagnosi solo in seguito a improvvisi e acuti problemi epatici, spesso nel contesto di un'anemia emolitica (anemia dovuta alla distruzione eccessiva dei globuli rossi). Ciò conduce a un'anomala produzione delle proteine e a difetti del metabolismo del fegato. Tale metabolismo deviato conduce a un accumulo di prodotti di scarto, come ammoniaca, nel circolo sanguigno. Se tale prodotto irrita il cervello, il paziente sviluppa un'encefalopatia epatica (che può condurre al coma, sino al pericoloso edema cerebrale).[6] L'anemia causa pallore, affaticabilità, tachicardia, dispnea da sforzo, ittero e alterata colorazione delle urine. L'esito finale è un'insufficienza epatica acuta o cronica grave.
Sintomi neuropsichiatrici
modificaCirca la metà dei pazienti con il morbo di Wilson presenta problemi neurologici o psichiatrici. La maggior parte dei pazienti inizialmente manifesta un deterioramento lieve reversibile delle capacità cognitive, assieme a cambiamenti comportamentali. In seguito giungono sintomi neurologici specifici, spesso sotto forma di parkinsonismo[21]: aumentata rigidità, ipertono, in casi gravi camptocormia, ossia tipica postura in avanti con arti in flessione, tratto di Gowers/festinatio e rallentamento dei comuni movimenti, con o senza un tipico tremore alle mani a riposo, scialorrea, espressioni facciali mascherate (amimia); comuni sono i difetti nell'articolazione del linguaggio (disartria); sono presenti anche atassia (assenza di coordinazione) e distonia temporanea, cioè improvvisi e incontrollati movimenti di parti del corpo: si notano contrazioni muscolari involontarie[22][23] simili a cloni e altre discinesie come posture bizzarre, estensioni forzate, torsioni attorno a una singola articolazione, crampi, spasmi, contratture, tremore[24] in movimento, in genere del capo o degli arti superiori, ipocinesia, miotonia, sindrome delle gambe senza riposo/acatisia.[25]
I sintomi neurologici più comuni nel morbo di Wilson sono tuttavia convulsioni ed emicrania.[6]
A volte si manifestano difficoltà a deglutire (disfagia), diplopia, affaticamento muscolare e generale, astenia, dolore muscolare, disautonomia, sincope, difficoltà nella scrittura (disgrafia e riduzione di dimensione della scrittura, cosiddetta micrografia parkisoniana).[26] Raramente può anche presentarsi con dei sintomi di apparente poli-neuropatia.[27]
Problemi psichiatrici dovuti alla malattia di Wilson possono includere cambi comportamentali quali la depressione, l'ansia, la confusione mentale, fino alla psicosi (delirio, paranoia, allucinazioni, potendo mimare la schizofrenia e il disturbo schizoaffettivo) e in fase avanzata sintomi somiglianti a quelli iniziali della demenza subcorticale e della demenza frontotemporale (disturbi dell'umore di origine non psichiatrica, impulsività, compromissione del giudizio, tendenza a promiscuità e ipersessualità oppure apatia e iposessualità, disfunzione esecutiva con scarsa pianificazione e processo decisionale, lentezza di pensiero, perdita di memoria ma senza segni di afasia, aprassia[28] o agnosia[29] presenti nelle vere demenze).[6] Si può infine verificare la paralisi pseudobulbare.
I sintomi psichiatrici sono comunemente osservati insieme con i sintomi neurologici, raramente si manifestano da soli. Questi sintomi spesso non sono ben definiti e si possono attribuire ad altre cause. Per questo la diagnosi del morbo di Wilson è raramente fatta quando solo i sintomi psichiatrici sono presenti, in quanto il medico può essere tratto in inganno pensando a una patologia puramente psichica.[21]
Altri organi
modificaDiversi organi sono coinvolti dall'accumulo di rame nella malattia di Wilson:[30]
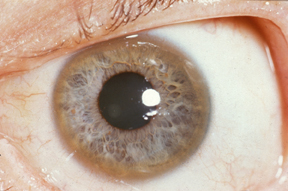
- occhi: anelli di Kayser-Fleischer. Essi sono dovuti alla deposizione di rame nella membrana di Descemet della cornea. Non si verificano in tutte le persone e possono essere visibili all'esame con la lampada a fessura. La malattia di Wilson è anche associata a cataratte, pigmentazione marrone o verde della capsula. Gli anelli di Kayser-Fleischer si verificano nel 66% dei casi;[20]
- reni e apparato scheletrico: acidosi tubulare renale di tipo "distale", una malattia che porta a nefrocalcinosi (nefropatia metabolica da accumulo di calcio nei reni), indebolimento delle ossa (a causa della perdita di calcio e fosfato) con osteoporosi, osteomalacia e artrosi (presenza di artralgia, osteofiti, artrosi vertebrale della colonna spinale che causa danni alle radici nervose)[31][32], e occasionalmente aminoaciduria (perdita di aminoacidi nelle urine, necessari per sintesi proteica);[6]
- cuore: la cardiomiopatia (debolezza miopatica del muscolo del cuore) acquisita è un problema raro, ma riconosciuto nella malattia di Wilson e può portare a insufficienza cardiaca e aritmie cardiache (episodi di irregolare battito del cuore);[6]
- ormoni e sistema endocrino: ipoparatiroidismo (insufficienza di funzionamento delle ghiandole paratiroidi, che porta a bassi livelli di calcio plasmatico), infertilità, amenorrea, pubertà ritardata e aborti spontanei abituali; raramente ipotiroidismo;[6][33]
- cute: a volte lunule delle unghie di colore ceruleo, xerosi (pelle secca), pigmentazione anomala, facilità alle infezioni cutanee;[34]
- miscellanea: anoressia, inappetenza, perdita del gusto (ageusia), secchezza e diradamento dei capelli (lieve alopecia), agranulocitosi, disturbi gastrointestinali e digestivi in conseguenza dei problemi al fegato, disturbi alle mucose, facilità all'anafilassi allergica.[35]
Genetica
modificaIl gene responsabile della malattia di Wilson (ATP7B) è stato individuato e mappato sul cromosoma 13 (13q14.3) e si esprime principalmente nel fegato, reni, e nella placenta. Il gene codifica per un'ATPasi di trasporto di tipo P che ha la funzione di regolare l'efflusso di rame dall'epatocita nella bile e il suo legame alla ceruloplasmina.[6] Le mutazioni sono rilevate nel 90% dei soggetti. La maggior parte (60%) sono mutazioni omozigoti del gene ATP7B (due copie anomale) e il 30% ha solo una copia anomala (mutazione eterozigote). Il dieci per cento non presenta alcuna mutazione rilevabile.[20]
Anche se 300 mutazioni del gene ATP7B sono state descritte, nella maggior parte dei casi di malattia di Wilson nella popolazione sono dovuti a un piccolo numero di mutazioni specifiche per quella data popolazione. Per esempio, nelle popolazioni occidentali, la mutazione del gene H1069Q (sostituzione di una istidina con una glutammina nella posizione 1069 della proteina) è presente nel 37-63% dei casi, mentre in Cina questa mutazione è molto rara, mentre la R778L (arginina al posto di leucina nella posizione 778) si trova più frequentemente. Relativamente poco si sa circa l'impatto relativo delle varie mutazioni, anche se la mutazione H1069Q sembra, secondo alcuni studi, portare a insorgenza soprattutto di problemi neurologici successivamente.[6][36]
Una variazione nel gene normale PRNP può modificare il decorso della malattia, ritardando l'età di insorgenza e modificandone i sintomi. Questo gene, infatti, produce una proteina prionica, che è attiva nel cervello e in altri tessuti e sembra anche essere coinvolta nel trasporto del rame.[37] Il ruolo del gene ApoE è stato inizialmente sospettato ma non è stato confermato successivamente da studi.[36]
La malattia è ereditata con modalità autosomica recessiva. Per la trasmissione, entrambi i genitori di un individuo affetto devono possedere un gene alterato. La maggior parte non ha una storia familiare della malattia.[36] Le persone con un solo gene anomalo sono chiamati portatori (eterozigoti) e possono avere lievi anomalie, clinicamente insignificanti, del metabolismo del rame.[38]
La malattia di Wilson è la più comune causa di un gruppo di malattie ereditarie che causano il sovraccarico di rame nel fegato. Tutte possono causare cirrosi in giovane età. Le altre malattie del gruppo sono: la cirrosi infantile indiana, la cirrosi tirolese infantile endemica e la tossicosi idiopatica del rame. Queste malattie non sono, però, legate a mutazioni del gene ATP7B. Infatti, per esempio, la cirrosi infantile indiana è stata collegata a mutazioni del gene KRT8 e KRT18.[36]
Fisiopatologia
modificaIl rame è necessario all'organismo per un certo numero di funzioni fisiologiche, esso è soprattutto un cofattore per il corretto funzionamento di una serie di enzimi, come la citocromo-c ossidasi, la ceruloplasmina, la dopamina β-idrossilasi, la superossido dismutasi e la monofenolo monoossigenasi.[36]
Il rame entra nel corpo attraverso il tratto digestivo. Una proteina trasportatrice:
- la CMT1, posta sulle cellule del piccolo intestino, trasporta il rame al loro interno, dove viene legato, in parte con qualche metallotioneina e in parte trasportato dalla ATOX1 verso l'apparato del Golgi dell'enterocita. Qui, in risposta alle concentrazioni crescenti di rame, un enzima chiamato ATP7A rilascia il rame nella vena porta e poi verso il fegato.
- le cellule epatiche possiedono anch'esse loro la proteina CMT1 che trasporta al loro interno il rame, dove esso viene legato da una proteina ad attività enzimatica ferrossidasica, la ceruloplasmina. Qui, l'enzima ATP7B, provvede a rilasciarlo nel sangue, oppure a rimuovere dal fegato quello in eccesso, secernendolo all'interno della bile.
Entrambe le funzioni di ATP7B risultano alterate nella malattia di Wilson. Il rame si accumula così nel tessuto epatico, la ceruloplasmina viene ancora secreta ma in una forma deficitaria di rame e che si degrada rapidamente nella circolazione sanguigna.[36]
Quando la quantità di rame nel fegato travolge le proteine che normalmente lo legano, si verifica il danno ossidativo attraverso un processo noto come "reazione di Fenton". Questa ossidazione patologica comporta un verificarsi di epatiti non infettive, fibrosi (deposizione di tessuto connettivo) e cirrosi. Il fegato rilascia nel sangue anche il rame che non è legato alla ceruloplasmina. Questo rame libero si diffonde in tutto il corpo ma va a colpire soprattutto gli occhi, i reni e il cervello.
Nel cervello la maggior parte del rame si deposita nei gangli basali, in particolare nel putamen e nel globo pallido (insieme chiamati nucleo lenticolare). Queste aree, è noto che normalmente partecipano al coordinamento del movimento e giocano un ruolo significativo nei processi neurocognitivi, come l'elaborazione degli stimoli e la regolazione dell'umore. Danni a queste aree (assieme alla possibile encefalopatia epatica), conseguentemente, producono i sintomi neuropsichiatrici osservati nella malattia di Wilson.[36]
Non è chiaro perché la malattia di Wilson causi emolisi, ma diversi indizi suggeriscono che alti livelli di rame non legato alla ceruloplasmina abbiano: un effetto diretto sull'ossidazione dell'emoglobina, sull'inibizione dei fornitori enzimatici di energia del globulo rosso o comportino un danno diretto sulla loro membrana cellulare.[39]
Diagnosi
modificaLa malattia di Wilson può essere sospettata sulla base di uno qualsiasi dei sintomi di cui sopra oppure quando un parente stretto ne è stato trovato affetto. La maggior parte dei pazienti presenta test di funzionalità epatica anomali, come elevate transaminasi e bilirubina. Se il danno epatico è significativo, i livelli di albumina possono essere bassi a causa di una incapacità delle cellule epatiche danneggiate di produrre questa proteina. Allo stesso modo, il tempo di protrombina (un test di coagulazione del sangue) può essere prolungato, in quanto il fegato non è in grado di produrre proteine note come fattori della coagulazione.[6] I livelli di fosfatasi alcalina sono relativamente bassi nei soggetti ammalati e questo è legato all'insufficienza epatica acuta.[40]
Non esiste un test completamente affidabile per la diagnosi del morbo di Wilson, ma i livelli di ceruloplasmina e rame plasmatici, nonché della quantità di rame escreto nelle urine durante un periodo di 24 ore, sono utilizzati per capire la quantità di rame presente nel corpo. La prova standard ideale per la diagnosi è comunque la biopsia epatica.[6]
Ceruloplasmina
modificaI livelli di ceruloplasmina sono anormalmente bassi (<0,2 g/L) nell'80-95% dei casi di malattia.[6] Possono, tuttavia, essere presenti livelli normali in soggetti con infiammazione in corso, in quanto è una proteina di fase acuta. Valori bassi di ceruloplasmina si trovano anche nella malattia di Menkes e nella aceruloplasminemia, malattie molto più rare della malattia di Wilson.[6][38]
Ferritina
modificaLa ferritina risulta essere alta.[41]
Anelli di Kayser-Fleischer
modificaLa combinazione di sintomi neurologici, livello basso di ceruloplasmina e la presenza degli anelli di Kayser-Fleischer negli occhi sono considerati sufficienti per la diagnosi del morbo di Wilson. In molti casi, tuttavia, sono necessari ulteriori accertamenti.[38]
Rame nell'urina e nel siero
modificaI valori di rame nel siero sono bassi, ma paradossalmente sono elevati nelle urine. Per l'esame, l'urina viene raccolta per 24 ore in una bottiglia con un rivestimento privo di rame. Livelli superiori a 100 µg/24h (1,6 µmol/24h) confermano la malattia di Wilson e dei livelli superiori a 40 µg/24h (0,6 µmol/24h) sono fortemente indicativi.[6] Alti livelli di rame nelle urine non sono però univoci per la malattia di Wilson. A volte essi vengono, infatti, osservati nelle epatiti autoimmuni e nelle colestasi (qualsiasi malattia che ostacola il flusso della bile dal fegato all'intestino tenue).[38]
Nei bambini, può essere utilizzato il test alla penicillamina. Viene somministrata una dose orale di 500 mg di penicillamina e vengono raccolte le urine delle 24 ore. Se queste contengono più di 1 600 mg (25 micromoli) di rame, ciò diventa un indicatore affidabile della malattia di Wilson. Questo test non è stato però convalidato per l'utilizzo su persone adulte.[38]
La biopsia epatica
modificaUna volta che gli esami hanno indicato il morbo di Wilson, il test ideale di certezza della diagnosi è il prelievo di una piccola quantità di tessuto epatico attraverso una biopsia epatica. Il tessuto prelevato viene valutato al microscopio per identificare il grado di steatosi epatica e cirrosi. Esami istochimici sono poi utilizzati per quantificare la quantità di rame e misurare la gravità della accumulo. Un livello di 250 µg di rame per grammo di tessuto epatico secco conferma la malattia di Wilson. Occasionalmente si possono trovare bassi livelli di rame, in tal caso combinazione dei risultati della biopsia con tutti gli altri esami effettuati potrebbe ancora portare a una diagnosi formale di Wilson.[6]
Nei primi stadi della malattia, la biopsia mostra tipicamente steatosi (deposizione di materiale grasso), aumento di glicogeno nel nucleo e aree di necrosi (morte cellulare). Nella malattia più avanzata, i cambiamenti osservati sono molto simili a quelli osservati nelle epatiti autoimmuni, come l'infiltrazione di cellule infiammatorie, necrosi frammentarie e fibrosi (tessuto cicatriziale). Nella malattia avanzata, infine, la cirrosi è la conseguenza principale. In caso di insufficienza epatica acuta, la degenerazione delle cellule epatiche e la distruzione dell'architettura normale del tessuto porta a un contesto cirrotico. I metodi istochimici per la rilevazione di rame sono incoerenti e inaffidabili e se effettuati da soli sono considerati insufficienti per stabilire una diagnosi precisa.[38]
Risonanza magnetica e/o TAC
modificaSe sono presenti sintomi neurologici, la risonanza magnetica del cervello viene di solito eseguita; essa può mostrare anche il "volto del panda gigante" caratteristico della malattia. La risonanza magnetica nucleare (RMN) e/o la tomografia assiale computerizzata (TAC) possono mostrare alterazioni nei gangli basali e nella materia bianca subcorticale, o atrofia di alcune parti di essi.[42]
I test genetici
modificaL'analisi delle mutazione del gene ATP7B, così come degli altri geni legati all'accumulo di rame nel fegato, può essere eseguita. Una volta che una mutazione è confermata è utile fare uno screening sui membri della famiglia.[6]
Diagnosi differenziale
modifica- Malattie neurodegenerative (es. malattia di Parkinson con demenza da corpi di Lewy, parkinsonismi da altra origine, altre demenze, corea di Huntington), neuromuscolari (es. MELAS) e demielinizzanti (es. sclerosi multipla)
- Malattia di Menkes, sindrome di Hallervorden-Spatz e aceruloplasminemia
- Corea di Sydenham
- Carenza di vitamina B12 (es. in celiachia, anemia perniciosa)
- Cirrosi epatica di altra origine (alcolismo, cirrosi infantile indiana, cirrosi tirolese infantile endemica, tossicosi idiopatica del rame)
- Patologie psichiatriche come schizofrenia, disturbo schizoaffettivo, psicosi...
- Nefropatie di altra origine
- Sindrome di Wilson (un disturbo legato all'ipotiroidismo).
Trattamento
modificaDieta
modificaIn generale, una dieta povera di alimenti contenenti rame è raccomandata, vanno evitati: funghi, noci, cioccolato, frutta secca, fegato animale e frutti di mare. Non è necessario, tranne in fase acuta di cirrosi, che il paziente escluda del tutto le bevande alcoliche, dato che non si tratta di un'insufficienza epatica primaria, ma dovuta a intossicazione cronica da rame.[6]
Farmacologico
modificaSono disponibili diversi trattamenti farmacologici per la malattia di Wilson. Alcuni tendono ad aumentare l'eliminazione del rame dal corpo mentre altri prevengono l'assorbimento del rame dalla dieta.
In generale, la penicillamina è il primo farmaco usato. Questa lega il rame (chelazione) e lo porta a essere escreto attraverso le urine. La penicillamina non è priva di controindicazioni: circa il 20%, infatti, soffre di effetti collaterali o complicanze del trattamento come il lupus farmaco-indotto (causa dolori articolari ed eruzioni cutanee). In coloro che hanno anche sintomi neurologici, la quasi metà soffre di un paradossale peggioramento dei sintomi. Mentre questo fenomeno si osserva anche in altri trattamenti per la malattia di Wilson, di solito questo è considerato come un'indicazione valida alla sospensione penicillaminica per incominciare un trattamento di seconda linea.[6][38] I pazienti intolleranti alla penicillamina possono incominciare la terapia con cloridrato di trientina (triethylene tetramine dihydrochloride o 2,2,2-tetramine), che possiede anche proprietà chelanti. Alcuni raccomandano la trientina come trattamento di prima linea, ma l'esperienza con penicillamina è maggiore.[38] Un agente con nota attività per il trattamento nella malattia di Wilson ulteriore è il tetratiomolibdate (tetrathiomolybdate); però questo trattamento è ancora considerato sperimentale,[38] anche se alcuni studi hanno mostrato un effetto benefico.[6]
Una volta che tutti i valori sono ritornati alla normalità, lo zinco (di solito sotto forma di una prescrizione di acetato di zinco, chiamato GALZIN) può essere utilizzato al posto di chelanti per mantenere stabili i livelli di rame nel corpo. Lo zinco stimola la metallotioneina, una proteina presente nelle cellule dell'intestino che lega il rame e impedisce il loro assorbimento e quindi il trasporto nel fegato. La terapia di zinco è continuata tranne quando avviene una ripresa dei sintomi o quando l'escrezione urinaria di rame aumenta.[38]
Nei rari casi in cui nessuno dei trattamenti per via orale risulti efficace e soprattutto nei casi di grave malattia neurologica, il dimercaprolo risulta necessario. Questo farmaco viene iniettato per via intramuscolare, ogni poche settimane, esso presenta una serie di spiacevoli effetti collaterali come il dolore muscolare, bruciore cutaneo, sintomi gastrointestinali, congiuntivite, sintomi rinologici, ipertensione arteriosa.[16]
Le persone che sono asintomatiche (ad esempio quelli a cui è stata diagnosticata grazie allo screening familiare o a seguito di test con risultati anomali) vengono generalmente trattate pure, poiché l'accumulo di rame può causare danni nel lungo termine. Non è chiaro se queste persone possano essere trattate meglio con la penicillamina o con l'acetato di zinco.[38]
La terapia fisica
modificaLa fisioterapia è utile per quei pazienti che presentano la forma neurologica della malattia. Il trattamento con chelanti del rame può richiedere fino a sei mesi per tornare a lavorare e la terapia fisica può aiutare a far fronte all'atassia, alla distonia e ai tremori, così come può impedire lo sviluppo di contratture che possono derivare da distonia.[43]
Trapianto
modificaIl trapianto di fegato è una cura efficace per la malattia di Wilson, ma è utilizzato solo in scenari di particolare gravità a causa dei numerosi rischi e complicazioni associate alla procedura chirurgica. Viene utilizzato principalmente nelle persone con insufficienza epatica fulminante che non rispondono al trattamento medico o in quelli con avanzata malattia epatica cronica. Il trapianto di fegato è evitato nei casi di grave malattia neuropsichiatrica, in cui la sua efficacia non è stata dimostrata.[6][38]
Note
modifica- ^ Il termine morbo, dal latino Morbus, "malattia che conduce a morte", è stato storicamente utilizzato per indicare le malattie a decorso fatale, soprattutto perché sconosciute e quindi incurabili. Attualmente è un vocabolo in via di abbandono sia per rispetto del malato, sia perché di molte malattie è stata trovata l'origine e la cura.
- ^ a b Malattia di Wilson su Osservatorio malattie rare
- ^ Malattia di Wilson - Fondazione Telethon
- ^ a b WB. Hu, YZ. Han; BC. Xue; N. Cheng; DY. Sun; DQ. Ye; RM. Yang, [Epidemiological study of hepatolenticular degeneration at Hanshan County, Anhui Province]., in Zhonghua Yi Xue Za Zhi, vol. 91, n. 13, aprile 2011, pp. 894-7, PMID 21600116.
- ^ a b Kinnier Wilson SA, Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver (PDF), in Brain, vol. 34, n. 1, 1912, pp. 295–507, DOI:10.1093/brain/34.4.295.
- ^ a b c d e f g h i j k l m n o p q r s t u v Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML, Wilson's disease, in Lancet, vol. 369, n. 9559, 2007, pp. 397–408, DOI:10.1016/S0140-6736(07)60196-2, PMID 17276780.
- ^ a b Robertson WM, Wilson's disease, in Arch. Neurol., vol. 57, n. 2, febbraio 2000, pp. 276–7, DOI:10.1001/archneur.57.2.276, PMID 10681092.
- ^ Cumings JN, The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration (PDF), in Brain, vol. 71, Dec, 1948, pp. 410–5, DOI:10.1093/brain/71.4.410, PMID 18124738.
- ^ McIntyre N, Clink HM, Levi AJ, Cumings JN, Sherlock S, Hemolytic anemia in Wilson's disease, in N. Engl. J. Med., vol. 276, n. 8, febbraio 1967, pp. 439–44, DOI:10.1056/NEJM196702232760804, PMID 6018274.
- ^ Cumings JN, The effects of B.A.L. in hepatolenticular degeneration, in Brain, vol. 74, n. 1, marzo 1951, pp. 10–22, DOI:10.1093/brain/74.1.10, PMID 14830662.
- ^ Denny-Brown D, Porter H, The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease), in N. Engl. J. Med., vol. 245, n. 24, dicembre 1951, pp. 917–25, DOI:10.1056/NEJM195112132452401, PMID 14882450.
- ^ Vilensky JA, Robertson WM, Gilman S, Denny-Brown, Wilson's disease, and BAL (British antilewisite [2,3-dimercaptopropanol]), in Neurology, vol. 59, n. 6, settembre 2002, pp. 914–6, PMID 12297577.
- ^ Walshe JM, Wilson's disease; new oral therapy, in Lancet, vol. 267, n. 6906, gennaio 1956, pp. 25–6, DOI:10.1016/S0140-6736(56)91859-1, PMID 13279157.
- ^ Walshe JM, Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride, in Lancet, vol. 1, n. 8273, marzo 1982, pp. 643–7, DOI:10.1016/S0140-6736(82)92201-2, PMID 6121964.
- ^ Harper PL, Walshe JM, Reversible pancytopenia secondary to treatment with tetrathiomolybdate, in Br. J. Haematol., vol. 64, n. 4, dicembre 1986, pp. 851–3, DOI:10.1111/j.1365-2141.1986.tb02250.x, PMID 3801328.
- ^ a b Walshe JM, Treatment of Wilson's disease: the historical background, in QJM, vol. 89, n. 7, luglio 1996, pp. 553–5, PMID 8759497.
- ^ Brewer GJ, Recognition, diagnosis, and management of Wilson's disease, in Proc. Soc. Exp. Biol. Med., vol. 223, n. 1, gennaio 2000, pp. 39–46, DOI:10.1046/j.1525-1373.2000.22305.x, PMID 10632959.
- ^ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW, The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene, in Nat. Genet., vol. 5, n. 4, 1993, pp. 327–37, DOI:10.1038/ng1293-327, PMID 8298639.
- ^ Tanzi RE, Petrukhin K, Chernov I, et al., The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene, in Nat. Genet., vol. 5, n. 4, 1993, pp. 344–50, DOI:10.1038/ng1293-344, PMID 8298641.
- ^ a b c Merle U, Schaefer M, Ferenci P, Stremmel W, Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study, in Gut, vol. 56, n. 1, 2007, pp. 115–20, DOI:10.1136/gut.2005.087262, PMC 1856673, PMID 16709660.
- ^ a b Lorincz MT, Neurologic Wilson's disease, in Annals of the New York Academy of Sciences, vol. 1184, 2010, pp. 173–87, DOI:10.1111/j.1749-6632.2009.05109.x, PMID 20146697.
- ^ Fahn S. The varied clinical expressions of dystonia. Neurol Clinics. 1984;2:541–554.
- ^ (EN) Dystonia - Dystonia Europe, in Dystonia Europe. URL consultato il 30 novembre 2016.
- ^ Roberto Erro, Ignacio Rubio-Agusti e Tabish A. Saifee, Rest and other types of tremor in adult-onset primary dystonia, in Journal of Neurology, Neurosurgery, and Psychiatry, vol. 85, n. 9, 1º settembre 2014, pp. 965–968, DOI:10.1136/jnnp-2013-305876. URL consultato il 29 novembre 2016.
- ^ M. C. Trindade, T. Bittencourt, G. Lorenzi‐Filho, R. C. Alves, D. C. de Andrade, E. T. Fonoff, E. Bor‐Seng‐Shu, A. A. Machado, M. J. Teixeira, E. R. Barbosa, G. G. Tribl, Restless legs syndrome in Wilson's disease: frequency, characteristics, and mimics, Acta neurologica scandinavica
- ^ La Malattia - Associazione Nazionale Malattia di Wilson ONLUS, su malattiadiwilson.org. URL consultato il 27 maggio 2019 (archiviato dall'url originale il 27 maggio 2019).
- ^ Keun-Hwa Jung, MD; Tae-Beom Ahn, MD; Beom S. Jeon, MD, PhD, Wilson Disease With an Initial Manifestation of Polyneuropathy, Jama Neurology
- ^ Incapacità di compiere movimenti volontari finalizzati a uno scopo o di comprendere l'uso di oggetti abituali, pur essendo integre l'intelligenza e la motilità
- ^ Disturbo della percezione caratterizzato dal mancato riconoscimento di oggetti, persone, suoni, forme, odori già noti, in assenza di disturbi della memoria e in assenza di lesioni dei sistemi sensoriali elementari.
- ^ Gaetano Crepaldi e Aldo Baritussio, Trattato di medicina interna, PICCIN, 2002, pp. 4687–, ISBN 978-88-299-1642-9. URL consultato il 27 agosto 2011.
- ^ MANAGEMENT OF PATIENT WITH HEPATOLIENAL SYNDROME - MANAGEMENT OF PATIENT WITH PORTAL HYPERTENSION - MANAGEMENT OF PATIENT WITH ASCITES, su intranet.tdmu.edu.ua. URL consultato il 28 maggio 2019 (archiviato dall'url originale il 14 maggio 2019).
- ^ Wilson Disease - Rare Disease
- ^ Jonathan D. Gitlina, Wilson disease - Clinical presentation, Journal of Gastroenterology
- ^ Seyhan M, Erdem T, Selimoğlu MA, Ertekin V., Dermatological signs in Wilson's disease. Pediatric International Official Journal of the Japan Pediatric Society, 2009 June
- ^ Definitions - Wilson's Disease Association, su wilsonsdisease.org. URL consultato il 28 maggio 2019 (archiviato dall'url originale il 28 maggio 2019).
- ^ a b c d e f g de Bie P, Muller P, Wijmenga C, Klomp LW, Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes, in J. Med. Genet., vol. 44, n. 11, novembre 2007, pp. 673–88, DOI:10.1136/jmg.2007.052746, PMC 2752173, PMID 17717039.
- ^ Grubenbecher S, Stüve O, Hefter H, Korth C, Prion protein gene codon 129 modulates clinical course of neurological Wilson disease, in Neuroreport, vol. 17, n. 5, 2006, pp. 549–52, DOI:10.1097/01.wnr.0000209006.48105.90, PMID 16543824.
- ^ a b c d e f g h i j k l Roberts EA, Schilsky ML, A practice guideline on Wilson disease (PDF) [collegamento interrotto], in Hepatology, vol. 37, n. 6, 2003, pp. 1475–92, DOI:10.1053/jhep.2003.50252, PMID 12774027.
- ^ GR Lee, Chapter 48: acquired hemolytic anaemias resulting from direct effects of infectious, chemical or physical agents, in Lee GR, Foerster J, Lukens J et al. (a cura di), Wintrobe's clinical hematology, vol 1, 10th, Williams & Wilkins, 1999, pp. 1298, ISBN 0-683-18242-0.
- ^ Shaver WA, Bhatt H, Combes B, Low serum alkaline phosphatase activity in Wilson's disease, in Hepatology, vol. 6, n. 5, 1986, pp. 859–63, DOI:10.1002/hep.1840060509, PMID 3758940.
- ^ Hisao Hayashi, Motoyoshi Yano, Yoshikazu Fujita, Shinya Wakusawa, Compound overload of copper and iron in patients with Wilson's disease, in Medical Molecular Morphology, vol. 39, 2006, pp. 121–126, DOI:10.1007/s00795-006-0326-7, PMID 16998622.
- ^ Das SK, Ray K, Wilson's disease: an update, in Nat Clin Pract Neurol, vol. 2, n. 9, settembre 2006, pp. 482–93, DOI:10.1038/ncpneuro0291, PMID 16932613.
- ^ Brewer GJ, Askari FK, Wilson's disease: clinical management and therapy, in Journal of Hepatology, vol. 42, Suppl 1, 2005, pp. 13–21, DOI:10.1016/j.jhep.2004.11.013, PMID 15777568.
Bibliografia
modifica- Joseph C. Segen, Concise Dictionary of Modern Medicine, New York, McGraw-Hill, 2006, ISBN 978-88-386-3917-3.
- Stephen L. Hauser, Harrison: Neurologia clinica, Casarile (Milano), McGraw-Hill, 2007, ISBN 978-88-386-3923-4.
- RE. Tanzi, K. Petrukhin; I. Chernov; JL. Pellequer; W. Wasco; B. Ross; DM. Romano; E. Parano; L. Pavone; LM. Brzustowicz, The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene., in Nat Genet, vol. 5, n. 4, dicembre 1993, pp. 344-50, DOI:10.1038/ng1293-344, PMID 8298641.
- (EN) George J. Brewer, Wilson's disease: a clinician's guide to recognition, diagnosis, and management, Springer, 1º maggio 2001, ISBN 978-0-7923-7354-4.
- (EN) Frederick J. Suchy, Ronald J. Sokol e William F. Balistreri, Liver disease in children, Cambridge University Press, 2007, pp. 639–, ISBN 978-0-521-85657-7.
- (EN) John McDonald, Andrew Burroughs e Brian Feagan, Evidence-Based Gastroenterology and Hepatology, John Wiley and Sons, 28 settembre 2010, pp. 493–, ISBN 978-1-4051-8193-8.
- (EN) Anthony S. Fauci, Eugene Braunwald, Dennis Kasper, Stephen Hauser, Dan L. Longo, Harrison's Manual of Medicine, McGraw Hill Professional, 19 marzo 2009, pp. 975–, ISBN 978-0-07-147743-7.
- (EN) Alex J. Mitchell, Neuropsychiatry and behavioural neurology explained[collegamento interrotto], Elsevier Health Sciences, 2004, pp. 171–, ISBN 978-0-7020-2688-1. URL consultato il 12 giugno 2011.
- (EN) Eugene R. Schiff, Michael F. Sorrell e Willis C. Maddrey, Schiff's diseases of the liver, Lippincott Williams & Wilkins, 2007, pp. 1032–, ISBN 978-0-7817-6040-9.
- (EN) Frances Talaska Fischbach e Marshall Barnett Dunning, A manual of laboratory and diagnostic tests, Lippincott Williams & Wilkins, 2009, pp. 640–, ISBN 978-0-7817-7194-8.
Linee guida
modifica- (EN) LM. Baddour, AE. Epstein; CC. Erickson; BP. Knight; ME. Levison; PB. Lockhart; FA. Masoudi; EJ. Okum; WR. Wilson; LB. Beerman; AF. Bolger, Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association., in Circulation, vol. 121, n. 3, gennaio 2010, pp. 458-77, DOI:10.1161/CIRCULATIONAHA.109.192665, PMID 20048212.
- (EN) NA. Khan, B. Hemmelgarn; RJ. Herman; CM. Bell; JL. Mahon; LA. Leiter; SW. Rabkin; MD. Hill; R. Padwal; RM. Touyz; P. Larochelle, The 2009 Canadian Hypertension Education Program recommendations for the management of hypertension: Part 2--therapy., in Can J Cardiol, vol. 25, n. 5, maggio 2009, pp. 287-98, PMID 19417859.
- (EN) GL. Myers, RH. Christenson; M. Cushman; CM. Ballantyne; GR. Cooper; CM. Pfeiffer; SM. Grundy; DR. Labarthe; D. Levy; N. Rifai; PW. Wilson, National Academy of Clinical Biochemistry Laboratory Medicine Practice guidelines: emerging biomarkers for primary prevention of cardiovascular disease., in Clin Chem, vol. 55, n. 2, febbraio 2009, pp. 378-84, DOI:10.1373/clinchem.2008.115899, PMID 19106185.
- (EN) DA. Fitzgerald, RJ. Massie; GM. Nixon; A. Jaffe; A. Wilson; LI. Landau; J. Twiss; G. Smith; C. Wainwright; M. Harris, Infants with chronic neonatal lung disease: recommendations for the use of home oxygen therapy., in Med J Aust, vol. 189, n. 10, novembre 2008, pp. 578-82, PMID 19012558.
- (EN) N. Beydon, SD. Davis; E. Lombardi; JL. Allen; HG. Arets; P. Aurora; H. Bisgaard; GM. Davis; FM. Ducharme; H. Eigen; M. Gappa, An official American Thoracic Society/European Respiratory Society statement: pulmonary function testing in preschool children., in Am J Respir Crit Care Med, vol. 175, n. 12, giugno 2007, pp. 1304-45, DOI:10.1164/rccm.200605-642ST, PMID 17545458.
- (EN) W. Wilson, KA. Taubert; M. Gewitz; PB. Lockhart; LM. Baddour; M. Levison; A. Bolger; CH. Cabell; M. Takahashi; RS. Baltimore; JW. Newburger, Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group., in J Am Dent Assoc, vol. 138, n. 6, giugno 2007, pp. 739-45, 747-60, PMID 17545263.
Voci correlate
modificaAltri progetti
modificaWikiversità contiene risorse su malattia di Wilson
Wikimedia Commons contiene immagini o altri file su malattia di Wilson
Collegamenti esterni
modifica- (EN) Wilson disease, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- http://spazioinwind.libero.it/claudioitaliano/wilson.htm
- http://www.aspe.vb.it/it/morbo_di_wilson.htm Archiviato il 26 novembre 2014 in Internet Archive.
- http://www.sanihelp.it/enciclopedia/scheda/7616.html
| Controllo di autorità | Thesaurus BNCF 50571 · LCCN (EN) sh85060301 · GND (DE) 4189925-8 · BNF (FR) cb124022187 (data) · J9U (EN, HE) 987007558058805171 · NDL (EN, JA) 00576079 |
|---|