Chimica forense/Tecniche analitiche
Test preliminari
[modifica | modifica sorgente]Una prima operazione da eseguire nel caso di alcune tipologie di prove forensi consiste nell'effettuare una serie di semplici procedure mirate a rilevare la presenza di un composto sospetto. Spesso tali procedure prevedono l'uso di test basati su reazioni chimiche che hanno il vantaggio di necessitare solamente di piccole quantità di campione. In un secondo momento verrà eseguita l'analisi vera e propria, ma in genere è il risultato dei test preliminari a suggerire il tipo di tecnica analitica più adatta per ottenere le informazioni di interesse circa il campione. L'analisi è preceduta anche dall'ottenimento della documentazione visiva riguardante le prove a disposizione. Questa viene acquisita attraverso tecniche basate su diverse sorgenti di luce che incrementano le informazioni ricavate visivamente dalla prova forense.
Test chimici
[modifica | modifica sorgente]I test chimici vengono utilizzati per identificare determinate sostanze in tempi brevi. La maggior parte dei test è basata su variazioni di colore oppure sul grado di solubilità della sostanza incognita. Il problema principale è rappresentato dal fatto che i test sono distruttivi nei confronti del campione a disposizione, il loro l'utilizzo è perciò consigliato nel momento in cui si dispone di una quantità sufficientemente elevata di campione. Quando possibile, questi test sono ampiamente utilizzati in quanto hanno il vantaggio di essere rapidi e diretti, a differenza di altre tecniche molto più complesse.
Nei test basati sui colori si impiega un reagente opportuno che, una volta aggiunto al campione, porta a un cambiamento di colore. Nel momento dell'analisi bisogna tenere conto della possibilità che il test fornisca un falso positivo o un falso negativo. Questo problema viene risolto con un'operazione di "taratura": si analizzano rispettivamente una soluzione contenente l'analita di interesse (controllo della positività) e un bianco in cui siamo certi che l'analita non sia presente (controllo della negatività).
Un altro metodo di screening è invece basato sulla solubilità: il campione viene introdotto in un recipiente, nel quale si aggiunge un solvente opportuno in quantità proporzionale alla quantità di campione (in genere il campione a disposizione è poco e quindi si utilizza poco solvente).
Densità
[modifica | modifica sorgente]
Un test preliminare molto semplice che permette di identificare diversi materiali consiste nella determinazione della densità (ρ). Questa è data dal rapporto tra la massa (m) di una sostanza e il suo volume (V):[1]
La densità viene misurata sfruttando il principio di Archimede secondo il quale un oggetto immerso in un fluido riceve una spinta dal basso verso l'alto uguale per intensità alla massa di fluido spostato.
Un campione forense può essere determinato qualitativamente e quantitativamente attraverso diversi metodi. Il "metodo del galleggiamento" è uno di questi e si basa sul fatto che un corpo solido con densità minore del liquido in cui è immerso tende a galleggiare. Viceversa, se è il liquido ad avere densità minore, allora il corpo affonda. Il corpo solido può anche rimanere in sospensione nel caso in cui abbia la stessa densità del liquido. Alternativamente si può impiegare un metodo basato sul "gradiente di densità": si utilizza un tubo riempito con una miscela di vari liquidi in diverse proporzioni (e quindi con densità differenti) che formano una serie di strati. La densità del singolo strato è fornita dalla formula seguente:[1]
Dove ρ è la densità della miscela, ρ1 è la densità del primo liquido, ρ2 è la densità del secondo liquido, V1 è il volume del primo liquido ed infine V2 è il volume del secondo liquido.
Nel tubo a gradiente viene poi introdotto il materiale solido, che tenderà ad affondare fino a quando non raggiungerà lo strato di liquido con la stessa densità. Per stimare la densità del campione incognito è necessario tarare il tubo inserendo al suo interno diversi solidi a densità nota.
Test con radiazioni luminose
[modifica | modifica sorgente]Un campione può essere analizzato anche attraverso metodi che prevedono l'utilizzo di radiazioni luminose, spesso con l'ausilio di un ingrandimento. Vengono utilizzate nella maggior parte dei casi radiazioni corrispondenti al visibile, ultravioletto (UV) e nell'intervallo dell'infrarosso (IR). Le informazioni sono registrate attraverso la fotografia che sfrutta la luce visibile e che permette di documentare i campioni forensi. Tuttavia, l'immagine può essere prodotta impiegando altre radiazioni dello spettro elettromagnetico come UV e IR che sono in grado di fornire altre informazioni circa il campione (per esempio la fotografia all'infrarosso permette di identificare sostanze non rilevabili nell'intervallo del visibile e quindi a occhio nudo).
Quando necessario è possibile migliorare le immagini sfruttando un particolare fenomeno noto come fotoluminescenza, che si verifica quando il campione si trova esposto a una determinata lunghezza d'onda. Quando una molecola interagisce con una radiazione sufficientemente energetica, può assorbirla e passare a uno stato eccitato; in seguito la molecola torna nel suo stato fondamentale attraverso una diseccitazione che può avvenire in diversi modi. L'energia in eccesso può essere dissipata attraverso l'emissione di una radiazione. In questo caso si avrà il fenomeno della fotoluminescenza. La fluorescenza è una forma di fotoluminescenza e coinvolge l'emissione derivante dallo strato meno eccitato di un singoletto allo stato fondamentale. Un altro esempio di fotoluminescenza è dato dalla fosforescenza, che riguarda l'emissione che segue al salto dal più basso stato eccitato di tripletto al più basso stato fondamentale di singoletto.[2]
Tecniche microscopiche
[modifica | modifica sorgente]Alcune proprietà del campione non sono osservabili a occhio nudo e in questo caso è opportuno utilizzare le cosiddette "tecniche microscopiche". Queste permettono di eseguire ingrandimenti di diversa entità sull'oggetto in esame, trovando la massima espressione con la microscopia elettronica, in grado di indagare a fondo la struttura del campione grazie all'elevatissimo grado di ingrandimento che la contraddistingue.
Microscopia ottica
[modifica | modifica sorgente]
Una della caratteristiche più vantaggiose del microscopio è la sua versatilità, in quanto si presta all'analisi di un'ampia gamma di campioni. Il microscopio ottico sfrutta il fenomeno di rifrazione della luce e permette vari ingrandimenti del campione utilizzando lenti differenti. L'analisi attraverso questo strumento fornisce informazioni di tipo chimico e allo stesso tempo permetto di osservare la struttura del campione. Esistono molte varianti del microscopio ottico, ad esempio i microscopi a comparazione, il microscopio stereoscopico, il microscopio a luce polarizzata e il microscopio a fluorescenza.
Le componenti principali di un microscopio ottico sono una sorgente luminosa, un condensatore, un obiettivo e un oculare. La radiazione luminosa proveniente dalla sorgente viene collimata dal condensatore e indirizzata verso campione. L'obiettivo è in grado di ingrandire l'immagine che arriva poi all'oculare con il quale è possibile regolare la messa a fuoco e ingrandire ulteriormente l'immagine. Per garantire una messa a fuoco efficiente e quindi una migliore immagine si utilizza un processo di allineamento, conosciuto come illuminazione di Köhler.[3]
Con il microscopio comparatore, due campioni posti l'uno di fianco l'altro vengono posti a confronto. Lo strumento è costituito dalla combinazione di due microscopi e attraverso un unico oculare è possibile osservare l'immagine fornita da entrambi i microscopi.
Il microscopio stereoscopico è in grado di offrire un'immagine tridimensionale del campione utilizzando la luce riflessa, e anch'esso si basa sull'accoppiamento di due microscopi. Trova la sua maggior applicazione nei test preliminari, in particolare quando è necessario un ingrandimento tra x2 e x100.[3]
Quando si analizzano campioni come fibre o minerali possono essere necessarie informazioni relative all'orientazione del campione: a tale scopo si impiega il microscopio a luce polarizzata (PLM, dall'inglese "polarizing light microscope"). Questo si compone di due filtri polarizzanti, di un polarizzatore posizionato sotto il ripiano porta campione e di un analizzatore posto sopra l'obiettivo (orientato perpendicolarmente rispetto al resto). Il PLM risulta essere molto utile quando si osservano materiali anisotropi (ovvero che presentano comportamenti diversi in funzione della direzione di osservazione).
Quando si vogliono studiare campioni interessati dal fenomeno della fluorescenza, la scelta migliore ricade sul microscopio a fluorescenza. Durante l'osservazione si utilizza una sorgente luminosa che emette nell'intervallo dell'ultravioletto. Il contributo della luce emessa per eccitazione viene corretto con filtri opportuni presenti nello strumento.
La preparazione del campione per l'analisi al microscopio dipende dalla natura del materiale e dal tipo di informazione che si vuole ottenere. Nel caso si usi un microscopio stereoscopico si possono evitare manipolazioni del campione. In alternativa si pone il campione su un vetrino e lo si ricopre con una pellicola oppure, nel caso si voglia osservare una sezione trasversale, lo si incorpora in una resina. Quando è necessario un ambiente umido si aggiunge al campione una goccia di glicerina. Può essere interessante conoscere l'indice di rifrazione di un determinato materiale e a tale scopo il campione viene immerso in un liquido con indice di rifrazione simile.
Microscopia elettronico a scansione
[modifica | modifica sorgente]
Attraverso il microscopio elettronico a scansione (SEM) la superficie del campione viene bombardata con un fascio di elettroni energetici, e l'immagine superficiale viene generata grazie all'interazione del fascio con la superficie e ai vari effetti che ne derivano: produzione di elettroni secondari, elettroni "backscattered" e la produzione di raggi X caratteristici. Per poter effettuare analisi elementari il SEM viene solitamente accoppiato con un rivelatore EDX ("energy-dispersive X-ray").
Il fascio di elettroni viene prodotto nel vuoto grazie a un cannone elettronico e, successivamente, convogliato in un unico punto tramite particolari lenti elettromagnetiche. Il fascio viene indirizzato sul campione e spostato lungo la superficie del campione, facendo in modo che gli elettroni superficiali vengono estratti per poi essere rivelati, amplificati e visualizzati.
Una fase molto delicata è rappresentata dalla preparazione del campione prima dell'analisi al SEM. Normalmente è necessario montare i campioni sopra un apposito disco metallico ("stub") utilizzando poi del nastro adesivo per far aderire correttamente il campione al disco. Questo metodo risulta problematico nel caso di campioni stratificati, quando è richiesta l'analisi di una superficie piana. In questo caso il campione viene tagliato longitudinalmente e la sezione trasversale viene levigata mediante l'uso di un abrasivo adatto come la pasta di diamante. Infine, il materiale viene incorporato in una resina per poi essere analizzato. Nel caso si voglia recuperare il campione è preferibile montarlo sopra lo stub per poi esporre la sezione trasversale al fascio di elettroni. Un metodo alternativo, definito "a scalini", prevede l'utilizzo di uno scalpello per esporre i diversi strati del campione.
Nel caso si voglia analizzare un materiale non-conduttore è necessario ricoprire la superficie con un film di materiale conduttore; in caso contrario, gli elettroni secondari prodotti dall'interazione con il fascio elettronico genererebbero un eccesso di cariche positive e l'immagine ottenuta dal microscopio risulterebbe sfocata. Il film superficiale è costituito da oro e carbone e viene applicato tramite un evaporatore sotto vuoto. Il materiale ricoprente non deve interferire con l'analisi elementare, per questo motivo deve essere selezionato opportunamente nel caso di un'analisi EDX. Per evitare di ricoprire il campione si può utilizzare un microscopio elettronico a scansione ambientale (ESEM, dall'inglese "environmental scanning electron microscopy"), il quale può essere utile anche nel caso di campioni instabili nel vuoto. L'ESEM rimuove gli eccessi di carica impiegando una pressione moderata di vapore acqueo (o in alternativa di un gas adeguato) nella camera contenente il campione.
Quando il campione interagisce con il fascio elettronico incidente vengono emessi gli elettroni secondari (SE). Questi forniscono informazioni relative alla topografia della superficie del campione. Quando gli elettroni vengono dispersi (diffusi) in modo elastico dagli atomi del campione in seguito all'interazione fascio-superficie si ottengono gli elettroni "backscattered" (BSE). L'immagine prodotta dai BSE è caratterizzata da una risoluzione peggiore rispetto a quella prodotta dagli SE ma ha il vantaggio di fornire informazioni circa la composizione del campione: le immagini corrispondenti a regioni contenenti atomi con numero atomico più elevato risultano essere più luminose. Il rivelatore usato per l'analisi di elementi costituenti è l'EDX in quanto fornisce uno spettro che indica la composizione relativa alle diverse regioni del campione.
Diffrazione di raggi X
[modifica | modifica sorgente]
La tecnica XRD (dall'inglese "X-ray diffraction") risulta molto utile per identificare gli atomi che compongono un campione solido e la loro disposizione nel reticolo cristallino. Si utilizza una radiazione (nell'intervallo dei raggi X) che sia quanto più simile alla distanza tra i piani del reticolo. In seguito all'interazione tra i raggi X e il reticolo verranno prodotti dei picchi di diffrazione di varia intensità. Si immaginino due radiazioni con angolo di incidenza θ che vengono diffratte mantenendo lo stesso angolo. Nel momento in cui la prima onda interagisce con il primo strato e la seconda con lo strato successivo del reticolo, le onde si definiscono in fase: si verifica un'interferenza costruttiva ad un angolo θ se la differenza di lunghezza del percorso è uguale ad un numero intero di lunghezze d'onda, nλ (dove n è un numero intero e λ è la lunghezza d'onda). Nella diffrazione di raggi X la relazione fondamentale è l'equazione di Bragg:[4]
dove d è è la distanza tra i piani atomici.
La tecnica XRD impiega una sorgente a raggi X corrispondente a un materiale in grado di emettere in questo intervallo e da un anodo che può essere costituito da cromo, ferro, rame o molibdeno. Il campione viene omogeneizzato per ottimizzare i risultati e introdotto come polvere. La quantità di campione necessaria è di pochi milligrammi, rendendo la tecnica molto interessante in ambito forense dove solitamente si ha a disposizione pochissimo campione. Durante l'analisi sul campione viene indirizzato un fascio di raggi X e si registra l'intensità dei raggi diffratti attraverso un detector montato su un carrello mobile, in grado di ruotare. La posizione angolare è misurata come 2θ (ovvero l'angolo di diffrazione). Il rivelatore si muove con velocità costante e un computer traccia l'intensità del raggio diffratto come funzione di 2θ.
Per una determinazione qualitativa del campione si utilizza il grafico ottenuto dall'analisi con XRD. Questo è specifico di ogni materiale: i picchi osservabili in un grafico XRD possono essere associati a una fase cristallina caratterizzata da una struttura specifica. Confrontando il grafico corrispondente all'analisi con altri conservati in un database è possibile determinare la tipologia del materiale in questione. Oltre a fornire informazioni di tipo qualitativo, quest'analisi, se eseguita con le dovute precauzioni, può anche permettere una quantificazione degli elementi. Uno svantaggio dell'XRD sta nel fatto che l'analisi qualitativa di miscele può essere complessa in quanto i picchi dei costituenti risultano spostati e tendono a sovrapporsi; questo problema può essere risolto attraverso il raffinamento di Rietveld, che permette di separare i singoli componenti di un grafico XRD.
Spettroscopia molecolare
[modifica | modifica sorgente]Le tecniche spettroscopiche si basano sull'analisi della radiazione elettromagnetica assorbita, emessa o diffratta dalle molecole o dagli atomi quando subiscono delle transizioni tra i livelli energetici. Le molecole e gli atomi esistono in stati discreti denominati livelli di energia. La frequenza ν della radiazione elettromagnetica associata ad una transizione tra due stati di energia ΔE è data dalla relazione:[5]
dove h è la costante di Planck.
Esistono diverse tecniche spettroscopiche che fanno riferimento alle radiazioni provenienti da diverse regioni dello spettro elettromagnetico per investigare la separazione dei livelli di energia delle molecole. Ad esempio, per ottenere informazioni inerenti alla struttura di un atomo si utilizza la spettroscopia atomica. La spettroscopia molecolare invece coinvolge anche transizioni rotazionali, vibrazionali e elettroniche. La spettroscopia molecolare può quindi essere utilizzata per caratterizzare un campione grazie alla frequenza spettrale associata ad ogni molecola.
Spettroscopia infrarossa
[modifica | modifica sorgente]

La tecnica della spettroscopia infrarossa è basata sulla vibrazione interna delle molecole. Lo spettro infrarosso viene ottenuto dall'interazione tra una radiazione infrarossa e il campione (o con la sua superficie) e la successiva determinazione della frazione di radiazione incidente assorbita ad una determinata energia. Alla frequenza della vibrazione di una parte della molecola corrisponde una banda in uno spettro di assorbimento. Per far sì che una una molecola possa assorbire nell'infrarosso deve possedere una caratteristica specifica: un momento di dipolo elettrico variabile durante la vibrazione. Questa caratteristica è conosciuta come regola di selezione per la spettroscopia infrarossa.
Lo spettrometro infrarossi a trasformata di Fourier (FTIR) è uno dei principali strumenti per la registrazione dello spettro infrarosso. La maggior parte degli strumenti viene impostata per registrare tra i 4000 e i 400 cm-1,[6] ovvero la regione di principale interesse. La radiazione uscente dalla sorgente attraversa un interferometro, producendo un segnale che può essere matematicamente trasformato per fornire rapidamente lo spettro richiesto. Prima di raggiungere il detector la radiazione interagisce col campione. Spesso la spettroscopia IR viene accoppiata con una tecnica di microscopia così da poter studiare campioni di dimensioni ridotte. La spettroscopia infrarossa ha il vantaggio di essere applicabile a un gran numero di campioni diversi. La spettroscopia a trasmissione, nella quale si misura l'assorbimento della radiazione dopo che questa attraversa campione, è il metodo tradizionale. Questa tecnica permette lo studio di solidi, liquidi e gas.
L'uso di tale tecnica è molto comune in ambito forense in quanto non è distruttiva nei confronti del campione ed è eseguibile anche fuori dal laboratorio. Nella spettroscopia a riflettanza totale attenuata (ATR) un cristallo viene fatto aderire alla superficie del campione. Nell'ATR la profondità della penetrazione della radiazione incidente sulla superficie del campione è una funzione della lunghezza d'onda, dell'indice di rifrazione del cristallo e dell'angolo della radiazione incidente. Il cristallo utilizzato in tale tecnica è fatto di un materiale che presenta un elevatissimo indice di rifrazione e scarsa solubilità in acqua. Fanno parte di questi materiali ZnSe, germanio e una miscela di bromuro e ioduro di tallio. Per analizzare campioni in polvere si utilizza la spettroscopia a riflettanza diffusa. La riflettanza diffusa è data dall'energia che penetra una o più particelle e viene riflessa in ogni direzione. I campioni possono essere analizzati tali e quali oppure miscelati con del KBr in polvere per aumentare la quantità di campione presente e quindi permettere l'analisi.[7]
Le informazioni presenti in uno spettro prodotto dalla spettroscopia IR sono la percentuale di trasmittanza, quella di assorbanza o quella di riflettanza come funzione della lunghezza d'onda. Ai modi in cui vibra il campione in oggetto è possibile associare le diverse bande dello spettro IR. La condizione per cui una molecola presenti assorbimento IR è che il momento di dipolo elettrico presente al suo interno cambi durante la vibrazione. La vibrazione può coinvolgere anche la lunghezza del legame o l'angolo di legame. Alcuni legami possono stirarsi in fase (stretching simmetrico) oppure fuori fase (stretching asimmetrico). Si individuano tre regioni principali di uno spettro IR: il lontano infrarosso (<400 cm-1), il medio infrarosso (4000-400 cm-1, il più utilizzato in ambito forense) ed il vicino infrarosso (13000-4000 cm 1).[6]
Spettroscopia Raman
[modifica | modifica sorgente]
Si tratta di un tipo di spettroscopia molto simile all'IR, solitamente viene utilizzata come sua complementare. La tecnica indaga il modo in cui vengono disperse le radiazioni da parte di un campione. Molte delle radiazioni disperse mantengono invariata la loro lunghezza d'onda (scattering di Rayleigh),[8] una piccola parte invece subisce un leggero incremento o decremento. Quando si registra un aumento delle lunghezze d'onda il processo è conosciuto come diffusione Raman Stokes, viceversa il processo prende il nome di diffusione Raman anti-Stokes. Lo scattering Raman delle molecole coinvolge le transizioni tra stati rotazionali o vibrazionali. Si deve avere un cambiamento in termini di polarizzabilità di un componente della molecola perché si verifichi la rotazione o la vibrazione molecolare.
La spettrometria Raman usa una sorgente di radiazioni che possono trovarsi nella regione del vicino ultravioletto, nel visibile o nel vicino infrarosso. Per minimizzare il fenomeno indesiderato della fluorescenza occorre particolare attenzione nella scelta della sorgente: risulta conveniente una sorgente di radiazioni a più alta lunghezza d'onda (nel vicino infrarosso). La radiazione luminosa dispersa proveniente dal campione attraversa una serie di lenti ottiche focalizzatrici e di raccolta. Si utilizza un filtro ottico per respingere la luce Rayleigh dispersa. La radiazione rimanente viene indirizzata verso il detector.
La microscopia Raman è una tecnica molto utile in chimica forense in quanto fornisce spettri con un'ottima risoluzione spaziale (dell'ordine di pochi micron). Solitamente si combina un microscopio con uno spettrometro. Il campione dopo essere stato posizionato nel porta-campione, viene illuminato da una luce bianca e, attraverso l'obiettivo, viene messo a fuoco. In seguito la lampada viene spenta e la radiazione proveniente dalla sorgente viene direzionata verso un separatore di lunghezze d'onda. La radiazione dispersa dal campione viene raccolta dall'obiettivo e inviata allo spettrofotometro. Esistono diverse tecniche di risonanza Raman, quali la spettroscopia Raman di risonanza (RRS) o la spettroscopia Raman amplificata da superfici (SERS).
Il vantaggio di questa tecnica consiste nel poter analizzare diversi tipologie di campioni con un pre-trattamento minimo. Per evitare di surriscaldare il campione con il laser della sorgente, lo si può raffreddare o lo si può porre in un'apposita cella rotante.
Spettroscopia UV-vis
[modifica | modifica sorgente]
La tecnica si basa sulle transizioni elettroniche associate all'assorbimento nell'UV (180-390 nm) e nel visibile (390-780 nm).[9] L'energia della radiazione associata a queste regioni è sufficiente a promuovere un elettrone esterno di una molecola presente in un dato livello di energia ad un livello energetico più alto. La transizione elettronica coinvolge una parte della molecola denominata cromofora. Il tipo di transizione risultante da un assorbimento UV-vis consiste nell'eccitazione di un elettrone dall'orbitale molecolare occupato più alto all'orbitale molecolare non occupato più basso.
Le componenti principali di uno spettrofotometro UV-vis sono: una sorgente di radiazioni, una cella contenente il campione, un elemento disperdente e un detector. Spesso si utilizzano due sorgenti luminose: una lampada a deuterio per la luce UV e una lampada a tungsteno per il visibile. Lo spettrometro a singolo raggio UV-vis è impostato in modo tale che il primo spettro a essere misurato sia quello della soluzione di riferimento, seguito poi dalla misura del campione di interesse. Nello spettrometro a doppio raggio le radiazioni vengono separate in due fasci paralleli, indirizzati attraverso due celle differenti: la prima contiene il solvente di riferimento, mentre la seconda il campione da analizzare. Tali celle sono solitamente fatte in quarzo o vetro, solitamente di lunghezza pari a 1 cm. Dal momento che in chimica forense ci si trova spesso a dover maneggiare quantità molto limitate di campione, questi vengono esaminati utilizzando un microspettrofotometro UV-vis (MSP).
Nella spettroscopia UV-vis solitamente si esaminano soluzioni diluite e l'intensità della radiazione trasmessa è funzione della concentrazione della molecola assorbente secondo la legge di Lambert-Beer:[10]
dove A è l'assorbanza della soluzione, c è la concentrazione, l è la lunghezza della cella contenente il campione ed infine ε è una costante che prende il nome di assorbività molare ed è caratteristica della molecola da analizzare.
Spettroscopia di fluorescenza
[modifica | modifica sorgente]
Il fenomeno della fluorescenza viene utilizzato in ambito forense in determinate tecniche spettroscopiche note con il nome di spettroscopia di fluorescenza, fluorimetria o spettrofluorimetria. Quando si ha fluorescenza, le collisioni molecolari fanno si che la molecola eccitata perda energia vibrazionale fino a raggiungere il livello vibrazionale più basso. Successivamente la molecola tende a rilasciare un fotone con energia pari alla differenza tra il livello vibrazionale più basso e lo stato fondamentale. La tecnica permette di analizzare molecole aromatiche o caratterizzate da atomi di carbonio coniugati. Nel fluorimetro l'angolo di emissione è di 90° rispetto alla direzione dell'eccitazione. La radiazione incidente sul campione viene selezionata mediante un monocromatore, mentre un secondo monocromatore è poi utilizzato per controllare la radiazione emessa dal campione. Questo lavora nel range che va dalla lunghezza d'onda di eccitazione fino alle lunghezze d'onda più elevate.
Lo spettro di emissione fornisce informazioni circa l'intensità di radiazione emessa in funzione delle varie lunghezze d'onda. La lunghezza d'onda di massima intensità è spesso utilizzata per scopi identificativi. Le molecole in grado di dare fluorescenza sono molte poche, ciò può rivelarsi contemporaneamente un vantaggio e uno svantaggio: da un lato è possibile analizzare matrici più o meno complesse sapendo che solo poche molecole saranno rilevabili limitando così eventuali interferenze, di contro la tecnica avrà un range di applicabilità piuttosto limitato.
Analisi elementare
[modifica | modifica sorgente]Nella disciplina forense l'identificazione e la quantificazione di elementi all'interno di diverse tipologie di prove sono aspetti estremamente importanti. L'analisi elementare permette di collegare un campione ad una scena del crimine o addirittura a chi l'ha commessa. La spettroscopia atomica comprende un insieme di tecniche molto sensibili che viene utilizzato per l'analisi di elementi: la tecnica prevede l'utilizzo di una fiamma, fornetto o plasma per decomporre il campione a livello atomico, determinando quindi le concentrazioni delle specie ottenute. Esistono diverse tecniche che permettono di eseguire le analisi elementari, come vedremo nei prossimi paragrafi.
Spettroscopia atomica
[modifica | modifica sorgente]

Le principali tecniche di spettrometria atomica usati in ambito forense sono la spettrometria di assorbimento atomico (AAS) e la spettrometria di emissione atomica (AES). Nell'AAS gli atomi assorbono una frazione della radiazione emessa dalla sorgente, mentre la radiazione non assorbita dal campione raggiunge il detector. Nell'AES la radiazione misurata deriva dalla diseccitazione degli atomi del campione, previa eccitazione ad opera della sorgente. Al centro di queste tecniche vi è la misurazione di emissione o assorbimento delle radiazioni a particolari lunghezze d'onda per registrarne lo spettro. AAS e AES garantiscono un mezzo di analisi per un'ampia gamma di elementi che possono essere determinati anche a bassissime concentrazioni, nell'ordine dei ppb o ppm.
In uno spettrometro di assorbimento atomico il campione viene atomizzato usando una fiamma (FAAS) o un fornetto di grafite (GFAAS). La FAAS ha il vantaggio di essere più economica se confrontata con la GFAAS, ma la fiamma richiede una quantità di campione più elevata (1-2 mL confrontati con i 5-10 μL della GFAAS).[11] La FAAS permette di determinare concentrazioni nell'ordine dei ppm,[11] mentre la GFAAS presenta una sensibilità maggiore riuscendo a rilevare e quantificare concentrazioni nell'ordine del ppb.[12]
La spettrometria di emissione atomica tradizionale usa la fiamma come mezzo di eccitazione degli atomi. Il campione viene iniettato come aerosol nella fiamma e l'intensità della radiazione emessa è misurata in corrispondenza della lunghezza d'onda selezionata. Gli elementi vengono identificati facendo riferimento alle linee di emissione prodotte. La tecnica AES tradizionale soffre di una ridotta sensibilità, per questo motivo si sostituisce la fiamma con una sorgente al plasma (ICP, dall'inglese "inductively coupled plasma"). Il plasma viene prodotto facendo fluire un gas carrier (tipicamente argon) il quale, con l'applicazione di un campo magnetico molto intenso, viene ionizzato. La temperatura del plasma può raggiungere i 10000 K,[12] permettendo di analizzare un range di elementi più ampio rispetto alla fiamma classica. La tecnica ha una sensibilità che si attesta nell'ordine dei ppb.
A livello operativo è necessario effettuare una calibrazione analizzando, oltre al campione, delle soluzioni standard esterne contenenti gli elementi di interesse. Nel caso dell'AAS, il valore di assorbanza è calcolato mediante l'intensità della radiazione trasmessa. I valori di assorbanza riferiti alle soluzioni standard vengono riportati in grafici di assorbanza in funzione della concentrazione, ottenendo una rappresentazione lineare per via della dipendenza dell'assorbanza nei confronti della concentrazione tramite la legge di Lambert-Beer. In alternativa si può utilizzare un metodo di calibrazione differente, definito "metodo delle aggiunte" che prevede l'aggiunta di standard a diverse concentrazioni (note) all'interno di varie aliquote di campione per poi misurare l'assorbanza. Nella spettrometria di emissione atomica l'intensità di emissione è proporzionale alla quantità di analita presente.
ICP-MS
[modifica | modifica sorgente]
La spettrometria di massa (MS) fornisce informazioni qualitative e quantitative di atomi, molecole o frammenti. Nell'MS gli analiti vengono ionizzati, e gli ioni risultanti vengono separati sulla base del loro rapporto loro massa/carica (m/z). La forma più usata dell'MS in ambito elementare è l'ICP. Tale combinazione permette di determinare metalli e alcuni non metalli a bassissime concentrazioni.
Nell'ICP-MS un campione liquido viene introdotto nello strumento sotto forma di aerosol utilizzando un gas carrier (argon o elio).[13] Il campione viene riscaldato e vaporizzato, portando poi all'atomizzazione ed infine alla ionizzazione. Solitamente questi strumenti utilizzano come detector un quadrupolo per separare gli ioni. Il metodo è quantitativo poiché il numero di ioni rivelati è direttamente proporzionale alla concentrazione di un dato elemento all'interno del campione. Solitamente il trattamento del campione prevede la solubilizzazione in un acido o una miscela di acidi (HNO3, HCl, HF ecc.).[13] Una strumentazione ICP-MS possiede un limite di rivelabilità (LOD, dall'inglese "Limit of Detection") dell'ordine del ppt.[14]
La tecnica ICP-MS può impiegare la laser ablation (LA) che consente di rimuovere del materiale (ablazione) da una piccola area di campione, iniettando quindi la parte rimossa tramite un flusso di gas carrier direttamente nel plasma. Il processo di ablazione è distruttivo, ma coinvolge un'area microscopica del campione, quindi viene normalmente utilizzato senza particolari problemi.
Il grafico che si ottiene da un'analisi ICP-MS vede l'intensità ionica in funzione di m/z (solitamente gli ioni hanno carica unitaria, quindi sull'assem delle ascisse viene riportata solo la massa). La tecnica si rivela estremamente utile per le analisi qualitative e quantitative multi-elementari, applicando un opportuno metodo di calibrazione (calibrazione esterna, metodo delle aggiunte, metodo dello standard interno o diluizione isotopica).
Spettroscopia di fluorescenza a raggi X
[modifica | modifica sorgente]
Quando si ha a che fare con poco campione, una tecnica non distruttiva come la spettroscopia di fluorescenza a raggi X (XRF) si rivela essere molto vantaggiosa per determinare la composizione elementare del campione senza doverlo distruggere. Il campione viene bombardato con un fascio di fotoni ad alta energia prodotti da un tubo radiogeno. La radiazione, interagendo con la materia, può essere assorbita da un atomo, trasferendo così tutta la sua energia ad un elettrone interno. Nel caso in cui la radiazione sia sufficientemente energetica, l'elettrone può venire espulso, creando una vacanza. La presenza di più vacanze causa l'instabilità dell'atomo; per tornare in una condizione di stabilità gli elettroni presenti negli strati più esterni vengono trasferiti negli strati più interni colmando le vacanze. Durante tale processo viene emesso una radiazione corrispondente all'intervallo dei raggi X che può dirsi caratteristica in quanto ha energia uguale alla differenza di energia dei due strati. Dal momento che ogni elemento contiene un set di energie unico, ognuno di loro produrrà un raggio X con un set di energie caratteristico.
I principali tipi di spettrometri XRF sono a dispersione di lunghezza d'onda (WDXRF, "wavelength-dispersive XRF") e a dispersione di energia (EDXRF, "energy-dispersive XRF"). Il primo misura la lunghezza d'onda, il secondo misura la radiazione di fluorescenza prodotta. Gli svantaggi principali della spettrometria WDXRF sono legati al costo dell'apparecchiature e alla quantità di campione relativamente grande necessaria all'analisi; ciò la rende una tecnica poco utilizzata nelle scienze forensi. La spettrometria EDXRF risulta essere più piccola e semplice, perciò è stato investito del tempo nel suo perfezionamento rendendola portatile e permettendo una rapida analisi del campione. Sono stati sviluppati anche spettrometri micro-XRF, in grado di analizzare piccolissime zone della superficie di campione.
I gusci elettronici coinvolti nella XRF solitamente sono quelli più interni (K e L).[15] Per produrre le linee K un elettrone dello strato L o M deve occupare una vacanza dello strato K, producendo un raggio X caratteristico e lasciando una vacanza nello strato L o M. Gli elettroni necessari a riempire le vacanze, interessati nella produzione di una linea L, appartengono agli strati M o N. Il raggio X prodotto viene chiamato K, L, M o N in base al tipo di guscio elettronico coinvolto nella transizione dell'elettrone. Si utilizzano poi le lettere α, β, γ per indicare da quale guscio elettronico proviene l'elettrone. Lo spettro che si ottiene dalla spettroscopia XRF mostra il numero come funzione dell'energia di legame; per identificare la sostanza si confronta lo spettro con uno standard. Si possono eseguire sia analisi quantitative che qualitative, con un LOD nell'ordine del ppm.[15]
Spettrometria di massa
[modifica | modifica sorgente]Quando si tratta di identificare un composto incognito la spettrometria di massa (MS) è una tecnica d'eccellenza. Infatti l'MS consente la caratterizzazione di molecole complesse anche quando si dispone di piccolissime quantità di campione. È possibile separare le tecniche MS in atomiche e molecolari.
Spettrometria di massa molecolare
[modifica | modifica sorgente]
L'MS offre informazioni riguardanti la massa delle molecole o di frammenti molecolari. In questa tecnica un campione allo stato gassoso viene bombardato con elettroni ad alta energia, provoca l'espulsione di uno o più elettroni. Gli ioni così prodotti vengono separati in funzione della loro massa applicando un campo magnetico. Il campione può essere introdotto direttamente (in tal caso può trovarsi anche allo stato solido) oppure tramite tecniche cromatografiche. Dopo l'introduzione del campione all'interno dello strumento, una sorgente di ionizzazione ne permette il passaggio in fase gassosa. In seguito le molecole vengono convertite in ioni e indirizzati verso un analizzatore di massa che li separa in base al loro rapporto m/z. Variando l'intensità del campo elettrico gli ioni vengono inviati a un detector moltiplicatore di elettroni. Quando si ha a che fare con campioni forensi i metodi di ionizzazione impiegati sono diversi a seconda del caso. La ionizzazione elettronica (EI) è il più comune e utilizza elettroni con energia di 70 eV.[16] Le molecole in tale processo tendono a perdere un elettrone producendo lo ione molecolare (M+). La ionizzazione chimica (CI)[16] consiste nel bombardare il campione con atomi carichi positivamente o con molecole invece che con elettroni. Tale metodo di ionizzazione è molto più soft rispetto all'EI, permettendo di analizzare sostanze impossibili da analizzare con l'altra tecnica. Una tecnica di ionizzazione particolarmente leggera è la ionizzazione elettrospray (ESI), nella quale il campione passa in fase gas in forma ionica attraverso un ago ad un potenziale di circa 4 kV.[16] Sono state inoltre sviluppate nuove sorgenti di ionizzazione che permettono di analizzare le superfici di campioni solidi senza effettuare prima una separazione o una preparazione. Queste tecniche sono la il desorbimento per ionizzazione elettrospray (DESI) e la direct analysis in real time (DART). Nella DESI viene prodotto un sottile film di liquido in cui l'analita viene disciolto nebulizzando goccioline cariche sulla superficie del campione, e il film viene successivamente portato nello spettrometro. Nella DART si genera un plasma ricco di atomi eccitati e ioni attraverso l'applicazione di un potenziale elettrico, e il plasma ad alta temperatura viene posto a contatto con la superficie del campione. Gli ioni degli analiti prodotti vengono desorbiti nella fase gas e trasportati all'interno dello spettrometro.
Per la spettrometria di massa sono disponibili diverse tipologie di analizzatori di massa. Tra questi c'è il quadrupolo, che prevede l'utilizzo di quattro barre alle quali viene applicata una differenza di potenziale lungo il percorso degli ioni. Quando a queste barre viene applicata una corrente diretta e una radiofrequenza gli ioni iniziano ad oscillare. A ogni rapporto m/z è possibile associare un'oscillazione stabile che permette agli ioni di viaggiare lungo tutto il cammino senza allontanarsi dalle barre. Applicando adeguatamente la differenza di potenziale è possibile selezionare solo gli ioni con un rapporto m/z voluto. La strumentazione a tempo di volo (TOF, dall'inglese "time of flight") sfrutta il fatto che ioni più leggeri vengono accelerati maggiormente rispetto a ioni pesanti, presentando quindi un "tempo di volo" minore lungo una specifica distanza. In uno spettrometro TOF gli ioni attraversano una regione senza alcun campo magnetico applicato e viene misurato il tempo necessario a raggiungere il detector. Lo spettrometro di massa può essere accoppiato con un altro spettrometro di massa (tandem MS, o MS-MS): il primo isola gli ioni desiderati a partire da una miscela, gli ioni di interesse vengono poi introdotti all'interno di un secondo spettrometro di massa dove vengono frammentati per produrre una serie di spettri.
Non sempre è possibile analizzare direttamente il campione forense, in tal caso sarà opportuno preparare il campione correttamente. Potrebbe essere necessario separare un analita dalla matrice (ad esempio i farmaci nelle analisi delle urine) in modo tale che la matrice non provochi interferenze. Nel caso in cui l'analita sia disciolto in un appropriato solvente si può optare per una semplice estrazione con solvente. Un altro tipo di estrazione utilizzabile è quella in fase solida (SPE, dall'inglese "solid phase extraction") che consiste nel separare gli analiti dalla matrice mediante l'uso di una fase stazionaria contenuta in una cartuccia. Un'altra opzione consiste nella pirolisi, una tecnica in cui il campione è riscaldato in maniera controllata per produrre un campione gassoso.
Lo spettro fornito in seguito a un'analisi allo spettrometro di massa rappresenta l'abbondanza relativa degli ioni rivelati in funzione del rapporto m/z. Il picco base corrisponde allo ione osservato più abbondante, al quale viene assegnato per convenzione un'abbondanza pari al 100%. Solitamente si esaminano ioni positivi, ma si possono analizzare anche quelli negativi. Nel caso in cui la sorgente di ionizzazione sia del tipo EI, il picco più intenso osservato nello spettro è associato allo ione molecolare (M+), ovvero la molecola di interesse che ha perso un solo elettrone formando una specie cationica radicalica. Ogni frammento ionico prodotto mostrerà un picco a valori di massa inferiori. Quando l'analita presenta un pattern di frammentazione ben conosciuto, si può utilizzare il SIM per identificarlo (Selected Ion Monitoring). Vengono selezionati diversi ioni come composto bersaglio per poi confrontare il loro rapporto con un standard. In alternativa si può usare una modalità "full scan" dove viene eseguito uno spettro di massa completo e viene successivamente comparato con quelli presenti in appositi database. Per poter eseguire delle analisi quantitative, bisogna utilizzare uno standard interno.
Spettrometria di massa a rapporto isotopico
[modifica | modifica sorgente]Gli isotopi si definiscono "stabili" quando non tendono a decadere attraverso processi radioattivi nel tempo. La maggior parte degli elementi presentano più di un isotopo stabile. La quantità di questi isotopi presenti in un determinato campione fornisce importanti informazioni riguardo alla sua origine, ad esempio nella determinazione della sorgente di tale materiale. La variazione nella composizione isotopica può essere misurata tramite uno spettrometro di massa a rapporto isotopico (IRMS).
Questa strumentazione utilizza diversi rivelatori allo scopo di misurare concentrazioni isotopiche piuttosto basse, in particolare per ogni isotopomero esiste uno specifico detector. I campioni solitamente vengono bruciati per ottenere gas semplici (CO2 e H2O) per condurre l'analisi. Dopo essere stato introdotto in una camera a ionizzazione, il gas viene accelerato applicando un campo magnetico. Gli ioni vengono indirizzati verso una coppa di Faraday adibita a raccogliere gli ioni con una determinata massa, registrando così la corrente ionica. Se ad esempio gli isotopi di interesse fossero quelli del carbonio bisognerà utilizzare tre coppe di Faraday, una per ciascun isotopo. Gli elementi e i corrispondenti isotopi di maggiore importanza nelle scienze forensi sono idrogeno (1H, 2H), carbonio (12C, 13C), azoto (14N, 15N), ossigeno (16O, 17O, 18O) ed infine zolfo (32S, 33S, 34S, 36S).[17] La tecnica IRMS può essere combinata con un gascromatografo per separare i componenti all'interno del campione.
Spettrometria di mobilità ionica
[modifica | modifica sorgente]La spettrometria di mobilità ionica (IMS) è stata perfezionata per la determinazione di tracce di gas e vapori. In tale tecnica un campione gassoso viene ionizzato e, applicando un campo elettrico mentre viaggiano a differenti velocità trasportati da un gas carrier, è possibile separare i differenti ioni presenti. Utilizzando una pompa si aspira il campione gassoso all'interno della camera di ionizzazione. I metodi di ionizzazione sono diversi, ma solitamente vengono impiegati dei radionuclidi (63Ni o 241Am) dal momento che sono caratterizzati da un'emivita lunga e che la strumentazione relativa richiede poca manutenzione. Successivamente gli ioni attraversano un tubo di deriva contenente il gas carrier e interagiscono con un campo elettrico a pressione atmosferica. Viene quindi rivelato il tempo di volo degli ioni. Nel caso di applicazioni forensi l'utilizzo di IMS portatili è molto popolare. Recentemente questa tecnica è stata combinata con la spettrometria di massa e/o con delle tecniche cromatografiche.
Tecniche separative
[modifica | modifica sorgente]Quando un chimico forense ha a che fare con una matrice molto complessa, riuscire a caratterizzare e quantificare le sostanze di interesse contenute al suo interno diventa particolarmente problematico. Le tecniche cromatografiche vengono in aiuto quando si tratta di dover affrontare questo tipo di problema separando gli analiti dalla matrice. Il principio generale su cui si basa la cromatografia è che su di una fase fissa detta fase stazionaria viene fatta scorrere un'altra fase (fase mobile). La fase stazionaria può essere può essere solida o liquida, supportata sulle pareti di una colonna o sulla superficie di particelle solide impaccate nella colonna.
Cromatografia su carta
[modifica | modifica sorgente]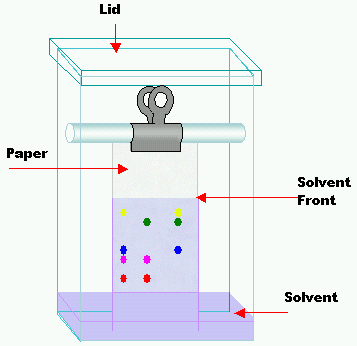
Si tratta della tecnica cromatografica più semplice in assoluto e, come si nota dal nome, il mezzo separativo in questione è proprio la carta. Questa è composta da cellulosa e funziona da fase stazionaria in quanto permette l'assorbimento delle molecole di acqua (polari). I solventi usati come fase mobile invece sono caratterizzati da polarità inferiore, solitamente composti da miscele di solventi organici e acqua. La carta viene inserita in un contenitore nel quale è presente un adeguato solvente, il quale inizierà a muoversi per capillarità attraverso la carta trasportando l'analita. L'entità del suo spostamento dipenderà dalla sua ripartizione tra fase stazionaria e fase mobile.
Per identificare un composto usando la cromatografia su carta si utilizzano i valori Rf. Tale valore si calcola nel seguente modo:[18]
La distanza percorsa è misurata in relazione al punto di deposizione e si effettua dal centro del punto. È da ricordare che le condizioni sperimentali influenzano l'Rf, quindi è importante conoscerle molto bene per poter confrontare i risultati.
Cromatografia su strato sottile
[modifica | modifica sorgente]
|

| |
TLC prima dell'eluizione
|
TLC dopo l'eluizione
|
Un metodo cromatografico semplice ed economico per l'analisi di campioni forensi è costituito dalla cromatografia su strato sottile, o TLC (dall'inglese "thin layer chromatography"). La fase stazionaria tipica è una lastrina e la miscela contenente il campione viene depositata ad un'estremità. La fase mobile è costituita da un solvente organico che scorre lungo il punto dove è stato deposto il campione. Il solvente, risalendo la lastrina per capillarità, permette la separazione delle varie sostanze nella miscela depositata. Infatti, ogni composto presente nella miscela del campione aderisce alla fase stazionaria e si solubilizza nel solvente in modo differente. La distanza percorsa in seguito all'eluizione è caratteristica di ogni composto.
La fase stazionaria ricopre uno strato sottile fatto da vetro o plastica. La deposizione del campione avviene in modo puntiforme su di una estremità della lastrina mediante un tubo capillare (la quantità depositata è importante in quanto quantità eccessive o limitate di campione non offrono buoni risultati). La lastrina viene successivamente posizionata in un contenitore al quale è stata aggiunta la fase mobile (ovvero una miscela di solventi adeguati). A fine eluizione, alla lastrina viene aggiunto un agente chimico adatto che, reagendo con i composti del campione, permette di rendere le macchie visibili alla luce UV.
Ovviamente risulta impossibile identificare un composto solo mediante il valore di Rf. Ma i valori di Rf di molti composti sono stati tabulati, per risalire al composto incognito è necessario compararlo con quelli conosciuti (si ricorda il discorso sull'importanza delle condizioni operative precedentemente fatto).
Gascromatografia
[modifica | modifica sorgente]
La tecnica della gascromatografia (GC) prevede l'introduzione di un campione gassoso o vaporizzato all'interno di una lunga colonna in grado di separare i costituenti del campione in base alle loro caratteristiche chimico-fisiche. I componenti vengono separati e fluiscono in sequenza dalla colonna verso il detector che identifica ogni composto misurando il suo tempo di ritenzione, ovvero il tempo necessario affinché un determinato composto esca dalla colonna e venga rivelato.
Attraverso un setto si inietta il liquido volatile o il campione gassoso in una zona riscaldata. Successivamente, con l'ausilio di un gas carrier (elio o idrogeno)[19] il vapore viene convogliato verso la colonna a temperatura controllata. I detector a disposizione sono diversi e vengono scelti in funzione del tipo di campione forense da analizzare. Il rivelatore a ionizzazione di fiamma (FID) permette di ionizzare l'analita con l'ausilio di una fiamma e la corrente risultante produce il segnale. L'analizzatore di energia termica (TEA) viene impiegato quando è necessario decomporre analiti contenenti azoto. Il rivelatore a cattura di elettroni (ECD) è un detector molto selettivo utilizzato per rivelare alogenuri o molecole a base di ossigeno.
Un tipo di accoppiamento estremamente vantaggioso è quello GC-MS; tale tecnica combinata permette una rapida identificazione dei componenti separati, cosa che la rende vantaggiosa e utilizzatissima in ambito forense. Dopo la separazione, il gas uscente dalla GC attraversa una camera di interfaccia necessaria a ridurne la pressione in modo da renderlo adatto alle condizioni di lavoro dell'MS.
Il campione deve essere opportunamente pre-trattato prima di poterlo analizzare, solitamente tramite derivatizzazione. Potrebbe essere utile eseguire una purificazione del campione prima della sua introduzione in colonna sfruttando tecniche come la SPE. La microestrazione in fase solida (SPME) permette invece di estrarre l'analita da un campione gassoso o liquido senza l'impiego di un solvente. In alternativa, quando la matrice risulta essere molto complessa, una tecnica efficace è costituita dalla pirolisi del campione.
In un cromatogramma il segnale prodotto dal detector è tracciato in funzione del tempo. Per identificare un determinato composto bisogna confrontare il suo tempo di ritenzione con quello presente in un database di composti ben noti. È possibile quantificare l'analita grazie all'area del picco che fornisce informazioni inerenti alla quantità di composto presente. Solitamente si utilizza uno standard interno (possibilmente un composto conosciuto che eluisca nei pressi dell'analita). Nella GC-MS l'approccio SIM permette di ottimizzare un'analisi di tipo quantitativo focalizzandosi solo su determinati picchi.
Cromatografia liquida
[modifica | modifica sorgente]
Come suggerisce il nome, la caratteristica principale che distingue questa tecnica dalla GC è il fatto che la fase mobile sia un liquido. Quando il campione da analizzare è instabile termicamente o non è volatile a sufficienza per essere adatto alla GC, la cromatografia liquida si rivela un valido sostituto.
Per ottimizzare la tecnica si lavora ad alte pressioni, in questo caso si parla di HPLC (cromatografia liquida ad alta prestazione), nella quale si forza un solvente a passare attraverso una colonna contenente particelle fini (5-10 μm di diametro).[20] Il solvente, il cui flusso è controllato per mezzo di una pompa, deve solubilizzare l'analita interessato. L'analisi si può eseguire in modalità isocratica (utilizzando lo stesso solvente, quindi a polarità fissa) oppure operando un gradiente di solvente (impiegando solventi diversi a polarità crescente).
La separazione del contenuto del campione analizzato avviene grazie alle differenti interazioni dei singoli composti con la fase stazionaria. Nella LC a fase normale la fase stazionaria è polare (composta da silice) e il solvente è apolare (ad esempio l'esano), e tale configurazione è ottimale per l'analisi di sostanze apolari.[20] Nella LC a fase inversa la fase stazionaria è apolare (silice modificata opportunamente)[21] e il solvente è polare (ad esempio l'acqua).[20]
Al termine della colonna è posto un detector che rivela i composti separati durante la corsa. Il più comune tra questi è il diode array detector (DAD) il quale misura l'assorbanza degli analiti nel campo dell'UV-vis, con una sensibilità dell'ordine del nanogrammo. In campo forense sono spesso utilizzati anche altri detector come quello a fluorescenza e lo spettrometro di massa (LC-MS).
Il cromatogramma risultante dalla LC è molto simile a quello che si ottiene con la GC: ai singoli composti è possibile associare un picco come funzione del tempo di ritenzione all'interno della colonna. Normalmente il cromatogramma della GC presenta più stretti della LC. Si può utilizzare il tempo di ritenzione di una sostanza per poterla identificare, ma un'analisi qualitativa più accurata è garantita dalla tecnica combinata LC-MS.
Cromatografia ionica
[modifica | modifica sorgente]La cromatografia ionica (IC) sfrutta il fenomeno di attrazione tra gli ioni in soluzione e i siti carichi presenti sulla fase stazionaria, e trova quindi grande applicazione nella determinazione di specie cariche. Come fase stazionaria si utilizza solitamente una resina a scambio ionico caratterizzata da gruppi funzionali carichi i quali interagiscono trattenendo i gruppi presenti nel campione aventi carica opposta. Quindi uno scambiatore anionico carico positivamente interagirà con gli anioni mentre uno scambiatore cationico carico negativamente interagirà con i cationi. Per eluire i composti trattenuti si fa uso di un'eluizione a gradiente o eluizione a variazione di pH.
Le resine a scambio ionico sono fondamentalmente costituite da materiale amorfo (un esempio sono le resine copolimeriche composte da stirene e divinilbenzene), e lavorando sulle proporzioni tra i costituenti è possibile ottenere diversi gradi di reticolazione nella resina. I gruppi aromatici possono essere modificati per contenere gruppi -SO3- per produrre una resina a scambio cationico; in alternativa, si possono introdurre gruppi -NR3+ per avere una resina a scambio anionico.[22]
Misurando la conduttività della soluzione si può valutare la presenza di ioni all'interno della stessa. È possibile eliminare elettroliti interferenti presenti in soluzione mediante la cromatografia a scambio ionico con soppressione; in quella anionica, ad esempio, la soluzione viene fatta passare all'interno di un "soppressore" dove i cationi vengono sostituiti da H+ per convertire l'eluente in H2O.
Anche in questo caso, la risposta del detector è riportata in funzione del tempo di ritenzione all'interno della colonna. I picchi possono essere utilizzati per identificare le varie specie ioniche presenti nella soluzione per comparazione con i risultati ottenuti dagli standard. Misurando l'area del picco si possono ottenere informazioni sulla concentrazione dell'analita.
Elettroforesi capillare
[modifica | modifica sorgente]
L'elettroforesi è un metodo che sfrutta il fenomeno di migrazione degli ioni contenuti in una soluzione a causa dell'influenza di un campo elettrico. Applicando una differenza di potenziale agli elettrodi immersi in una soluzione tampone (contenente molecole di interesse analitico), gli ioni presenti nel campione migrano verso uno degli elettrodi. La velocità di migrazione è funzione della carica e delle dimensioni delle particelle. L'elettroforesi capillare (CE) utilizza un capillare per poter applicare un grande campo elettrico, garantendo così una miglior risoluzione e un minor tempo di analisi rispetto alle tecniche elettroforetiche tradizionali.
In questa tecnica viene applicata una differenza di potenziale dell'ordine dei 10-30 kV attraverso un capillare riempito da una soluzione tampone.[23] Il materiale tradizionale che costituisce i capillari è la silice fusa;inoltre i capillari presentano un volume molto piccolo. La migrazione degli ioni è solitamente rivelata tramite un detector UV-vis, a fluorescenza o conduttimetrico.
Esistono diverse tipologie di elettroforesi capillare, ma nelle analisi forensi la elettroforesi di zona capillare (CZE) e la cromatografia elettrocinetica micellare (MECC o MEKC) sono le più popolari. La CZE viene eseguita in una soluzione tampone e la separazione si basa sulla differenza di mobilità elettroforetica. La MEKC è molto utile per la separazione di specie neutre, altrimenti difficilmente separabili con altre tecniche elettroforetiche; essa sfrutta un tensioattivo che, aggiunto alla soluzione tampone, genera delle micelle in grado di "imprigionare" al loro interno le molecole neutre di analita. In questo modo la loro migrazione verso l'elettrodo risulta facilitata.
Nell'elettroforesi capillare il detector fornisce un elettroferogramma, il quale mostra i picchi relativi ai vari composti in funzione del tempo di migrazione. Paragonando questi tempi con quelli di soluzioni standard, si possono eseguire delle analisi qualitative.
Analisi termiche
[modifica | modifica sorgente]Attraverso le analisi termiche si osservano e misurano i cambiamenti fisici e chimici che interessano un determinato materiale quando questo viene riscaldato. Tali cambiamenti includono processi di decomposizione, il rilascio o l'assorbimento di energia o ancora l'aumento o la perdita di massa. Questi cambiamenti si verificano a determinate temperature, caratteristiche per ciascun materiale. I metodi termici più interessanti in ambito forense sono le tecniche di pirolisi, la calorimetria a scansione differenziale, l'analisi termica differenziale ed infine l'analisi termogravimetrica.
Tecniche di pirolisi
[modifica | modifica sorgente]La pirolisi è un metodo che si basa sul riscaldare una sostanza in atmosfera inerte portandola fino a temperature elevate. Come conseguenza di questo riscaldamento si ha la produzione di frammenti molecolari caratteristici del materiale di partenza. Il pirolizzatore viene accoppiato con un gascromatografo, uno spettrometro di massa o addirittura con entrambi, in modo da poter identificare correttamente questi prodotti.
I tipi di pirolizzatori disponibili più comuni sono tre:
- pirolizzatore a fornace, in cui il campione viene posto in un fornetto precedentemente scaldato;
- pirolizzatore a punto di Curie, in cui il campione viene posizionato all'interno di un filamento ferromagnetico all'interno di un campo a radiofrequenze;
- pirolizzatore a riscaldamento resistivo, che utilizza un filamento di platino resistente al calore per scaldare il campione.
I pirolizzatori richiedono quantità di campione nell'ordine del microgrammo e operano un rapido riscaldamento fino a temperature nel range di 600-800 °C.[24] Lavorare a condizioni operative ben definite è importantissimo in queste tecniche per renderle riproducibili, altrimenti si rischia di andare incontro a reazioni secondarie indesiderate.
A seguito della pirolisi il prodotto viene inviato al gascromatografo o allo spettrometro di massa. Nella pirolisi-gascromatografia (Py-GC) il prodotto di combustione viene separato e identificato con un gascromatografo. Il cromatogramma risultante prende il nome di pirogramma. La pyrolysis-capillary GC è una tecnica accoppiata molto più sensibile: si utilizza una colonna capillare invece che una impaccata così da poter garantire una miglior risoluzione. È possibile analizzare l'eluato proveniente dalla GC con uno spettrometro di massa, in tal caso la tecnica prende il nome di Pyrolysis GC-MS (Py-GC-MS). È altresì possibile accoppiare direttamente il pirolizzatore ad un spettrometro di massa, ma una combinazione di questo tipo è molto più di nicchia.
In seguito alla decomposizione termica si ottengono frammenti che dipendono dalla struttura molecolare della molecola di partenza e dalle condizioni termiche. Esistono infatti tantissimi percorsi di decomposizione, per i polimeri ad esempio alcune tipiche reazioni di pirolisi sono la depolimerizzazione (il polimero ritorna a monomero), la scissione dei gruppi laterali (i gruppi legati alla catena principale si rompono rendendo la catena principale insatura) e la scissione randomica della catena (la catena polimerica viene rotta in modo randomico).
Grazie a questo metodo si ottiene un pirogramma, caratteristico del campione, che può essere confrontato con quelli contenuti in apposite banche dati, permettendo l'identificazione della sostanza incognita. I pirogrammi solitamente sono complessi, quindi non si paragonano tutti i picchi ma si fa riferimento esclusivamente a quelli più intensi.
Calorimetria a scansione differenziale e analisi termica differenziale
[modifica | modifica sorgente]
Queste due tecniche sono molto utili per caratterizzare le proprietà chimico-fisiche di un materiale. La calorimetria a scansione differenziale (DSC) registra l'energia da fornire al campione in modo che la sua temperatura eguagli quella di un materiale di riferimento, riportando quindi i dati di energia fornita in funzione della temperatura o del tempo. Le due sostanze vengono riscaldate o raffreddate in ambiente controllato alle stesse condizioni operative. L'analisi termica differenziale (DTA), invece, misura la differenza di temperatura tra il campione e il materiale di riferimento. I dati sono riportati in funzione del tempo o della temperatura.
Una piccola quantità di campione, dell'ordine del milligrammo, viene posizionata in un crogiolo che a sua volta viene inserito all'interno di una fornace. Il materiale di riferimento impiegato solitamente è l'allumina (Al2O3). Per mantenere l'atmosfera della camera in uno stato controllato, questa può essere riempita con un gas adeguato. Dopo aver programmato la velocità di riscaldamento, la fornace viene scaldata elettricamente (tipicamente 10 °C min-1, ma si può arrivare fino a 100 °C min-1).[25] Si possono registrare anche temperature inferiori a quella ambiente utilizzando azoto liquido.
Il grafico ottenuto in seguito a un'analisi DSC riporta il flusso di calore come funzione della temperatura a velocità costante di riscaldamento. Si ottiene un cambiamento nella linea di base quando varia la capacità termica del campione. L'equazione alla base di questa tecnica è:[26]
Dove ΔT è la differenza di temperatura tra il campione e il materiale di riferimento, q è la velocità di riscaldamento, Cp è la capacità termica del campione ed infine K è il fattore di calibrazione dello strumento. Si può calcolare la variazione di entalpia grazie al calcolo dell'area compresa tra la curva e la linea di base.
DSC e DTA sono tecniche molto utilizzate in ambito forense per caratterizzare le proprietà termiche di materiali a base polimerica. Per tali campioni si osserva la temperatura di transizione vetrosa, quando viene raggiunta questa temperatura il polimero cessa di essere vetroso e assume caratteristiche gommose. Tale temperatura si può determinare mediante l'utilizzo della DSC, il segnale corrispondente è rappresentato da una variazione endotermica dalla linea di base. Sono molti i fattori che influenzano il valore di questa temperatura, ad esempio la natura dei sostituenti, la struttura copolimerica, i tipi di legami tra catene, il peso molecolare, la presenza di plastificanti ecc. La temperatura si registra nel momento in cui si osserva l'inizio della transizione e non all'apice del picco.
Un'altra informazione che viene normalmente raccolta è quella relativa alla temperatura di fusione, ovvero il range di temperature in cui un polimero cristallino si scioglie. Nel grafico della DSC si osserva un picco endotermico corrispondente a questo intervallo.
Analisi termogravimetrica
[modifica | modifica sorgente]
Nell'analisi termogravimetrica (TGA) l'informazione di interesse è quella relativa alla massa di materiale persa come funzione della temperatura. La TGA permette di quantificare la variazione di massa di un materiale quando questo viene interessato da processi degradativi. Differenti sostanze mostrano un unico schema di decomposizione, ma la TGA fornisce dei dati sulla massa persa caratteristici per ogni campione.
Il campione viene posizionato sul braccio di una bilancia estremamente sensibile, a sua volta posta all'interno di una fornace. La variazione della massa del campione viene registrata mentre il campione è mantenuto ad una specifica temperatura o è soggetto a una variazione programmata di temperatura. Si possono eseguire riscaldamenti da -196 °C a 2400 °C e si possono utilizzare atmosfere differenti (azoto, ossigeno, argon o elio).[27] La TGA si può accoppiare anche con altre tecniche spettroscopiche per poter aumentare il numero di informazione relative al campione. Le quantità di campione introdotte sono dell'ordine del milligrammo e possono essere sotto forma di polveri, liquidi o fibre.
Il grafico TGA riporta la massa persa come funzione della temperatura. Osservando un grafico si possono notare svariati step: il primo corrisponde all'evaporazione del solvente, il secondo al primo processo di degradazione, il terzo al secondo processo di degradazione e così via fino ad arrivare al residuo finale il quale non decompone nel range di temperature impostate. La derivata di tale curva (DTG) mostra i picchi relativi ad ogni processo di degradazione in modo da evidenziare i punti in cui si è registrata la maggiore perdita in termini di massa del campione.
Note
[modifica | modifica sorgente]- ↑ 1,0 1,1 Stuart, p. 32
- ↑ Stuart, p. 35
- ↑ 3,0 3,1 Stuart, p. 42
- ↑ Stuart, p. 62
- ↑ Stuart, p. 70
- ↑ 6,0 6,1 Stuart, p. 73
- ↑ Stuart, p. 74
- ↑ Stuart, p. 90
- ↑ Stuart, p. 95
- ↑ Stuart, p. 97
- ↑ 11,0 11,1 Stuart, p. 114
- ↑ 12,0 12,1 Stuart, p. 115
- ↑ 13,0 13,1 Stuart, p. 117
- ↑ Stuart, p. 118
- ↑ 15,0 15,1 Stuart, p. 120
- ↑ 16,0 16,1 16,2 Stuart, p. 130
- ↑ Stuart, p. 135
- ↑ Stuart, p. 144
- ↑ Stuart, p. 149
- ↑ 20,0 20,1 20,2 Stuart, p. 156
- ↑ Stuart, pag.156
- ↑ Stuart, p. 159
- ↑ Stuart, p. 161
- ↑ Stuart, p. 168
- ↑ Stuart, p. 171
- ↑ Stuart, p. 172
- ↑ Stuart, p. 176