O ciclo da urea é un ciclo de reaccións metabólicas que ten lugar en moitos animais, que produce urea ((NH2)2CO) a partir de amoníaco (NH3). O ciclo da urea permite que o amoníaco, procedente da desaminación dos aminoácidos, non se acumule no organismo, xa que é unha substancia tóxica[1]. Nos mamíferos, o ciclo da urea ten lugar fundamentalmente no fígado, e en pequena proporción noutros órganos como o riles. Este ciclo foi descuberto por Hans Krebs e Kurt Henseleit en 1932, e foi o primeiro ciclo metabólico en ser descuberto, cinco anos antes ca o ciclo do ácido cítrico, tamén descuberto por Krebs.
Os organismos que non poden eliminar o amoníaco de forma fácil e rápida xeralmente teñen que convertelo nalgunha outra substancia, como a urea ou o ácido úrico, que son moito menos tóxicos ca o amoníaco. Nalgúns trastornos xenéticos e nos casos de fallo hepático o ciclo da urea non funciona correctamente. O resultado pode ser a acumulación de residuos nitroxenados, principalmente amoníaco, o que causa encefalopatía hepática.
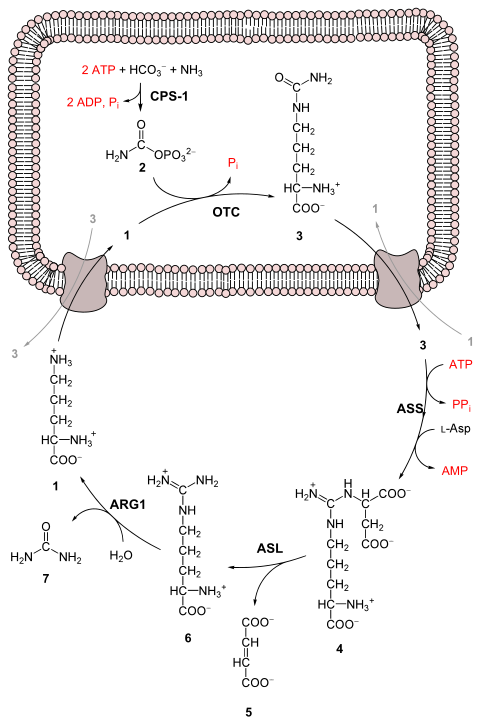
O ciclo da urea consta de cinco reaccións: dúas son mitocondriais e tres citosólicas. O ciclo converte dous grupos amino, un do NH4+ e outro do aspartato, e un átomo de carbono procedente do HCO3−, en urea, un produto de excreción relativamente pouco tóxico, para o que se gastan catro enlaces fosfato de "alta enerxía" (3 ATP hidrolizados a 2 ADP e 1 AMP). O transportador destes átomos de nitróxeno e carbono é o aminoácido non proteico ornitina, polo que ao ciclo tamén se lle chama ás veces ciclo da ornitina.
Pero como o fumarato se orixina cando o aspartato perde o seu grupo amino en forma de NH3 (por medio das reaccións 3 e 4), e como ademais PPi + H2O → 2 Pi, a ecuación global pode simplificarse así:
Téñase en conta tamén que as reaccións relacionadas co ciclo da urea tamén causan a oxidación de 2 NADH, polo que, en realidade, o ciclo da urea libera un pouco máis de enerxía da que consome. Estes NADH prodúcense de dúas maneiras:
Unha molécula de NAD+ redúcese a NADH polo encimaglutamato deshidroxenase durante a conversión do glutamato a amoníaco e α-cetoglutarato. O glutamato aquí funciona como o transportador non tóxico de grupos amino. Isto proporciona o ión amonio utilizado na síntese inicial de carbamoíl fosfato (tamén chamado fosfato de carbamoilo).
O fumarato liberado no citosol convértese en malato pola fumarase citosólica. Este malato é despois convertido en oxalacetato pola malato deshidroxenase citosólica, xerando outra molécula de NADH no citosol. O oxalacetato é un dos cetoácidos preferidos polas transaminases, e será reciclado a aspartato, mantendo o fluxo de nitróxeno no ciclo da urea.
Os dous NADH producidos poden fornecer enerxía para a formación de 5 ATP, o que supón unha produción neta dun enlace de alta enerxía por cada volta do ciclo da urea. Non obstante, se a gliconeoxénese esta activa no citosol, o último equivalente redutor vai utilizarse para impulsar á inversa o paso catalizado pola gliceraldehido-3-fosfato deshidroxenase en vez de xerar ATP.
A síntese de carbamoíl fosfato e o ciclo da urea dependen da presenza de N-acetilglutamato, que activa alostericamente a carbamoíl fosfato sintetase 1 (CPS1). A síntese de N-acetilglutamato pola N-acetilglutamato sintase (NAGS), está estimulada tanto pola arxinina, que é o estimulador alostérico da NAGS, coma polo glutamato, que é un produto das reaccións de transaminación e un dos substratos da NAGS, a cantidade de ambas as substancias aumenta cando se incrementa a de aminoácidos libres. Así, a arxinina non só é un substrato do ciclo da urea senón que serve tamén como activador do ciclo da urea.
O resto dos encimas do ciclo están regulados pola concentración dos seus substratos. As deficiencias hereditarias nos encimas do ciclo distintos da arxinase 1 (ARG1) non orixinan unha diminución significativa na produción de urea (a falta total de calquera dos encimas do ciclo orixina, polo contrario, a morte pouco despois do nacemento). O que ocorre máis ben é que se acumulan os substratos dos encimas deficientes, e incrementan a velocidade da reacción deficiente ata o normal.
Porén, a acumulación anómala de substrato ten tamén o seu custo. As concentracións de substrato son elevadas en todas as reaccións anteriores do ciclo ata o NH4+, o que ten como resultado unha hiperamonemia (incremento do [NH4+]P).
Aínda que a causa última da toxicidade do NH4+ non se comprende completamente, unha elevación forte das cantidades de [NH4+] supón un enorme estrés para o sistema de compensación do NH4+, especialmente para o cerebro humano (os síntomas das deficiencias no ciclo da urea inclúen atraso mental e astenia). Este sistema de compensación implica ás proteínas glutamato deshidroxenase 1 e glutamina sintetase, as cales fan decrecer as cantidades de oxoglutarato e glutamato. O cerebro é moi sensible á diminución destas substancias. A diminución de oxoglutarato fai descender a velocidade do ciclo do ácido cítrico, e o glutamato funciona como neurotransmisor e como precursor do GABA, outro neurotransmisor. [1](p.734)
As persoas con defectos xenéticos en calquera dos encimas que interveñen na formación de urea non poden tolerar unha dieta rica en proteínas. Os aminoácidos inxeridos en exceso con respecto aos requirimentos diarios mínimos para a síntese de proteínas desamínanse no fígado, producindo amoníaco libre, que non pode ser convertido en urea e exportado ao sangue, e o amoníaco é moi tóxico. Os humanos, porén, non podemos vivir cunha dieta sen proteínas. Non podemos sintetizar a metade dos 20 aminoácidos estándar, e estes aminoácidos esenciais deben ser achegados pola dieta.
Aos pacientes con defectos no ciclo da urea aplícanselles diversos tratamentos. A administración coidadosa dos ácidos aromáticos bencenoato ou fenilacetato na dieta pode axudar a reducir os niveis de amoníaco en sangue. O bencenoato convértese en bencenoíl-CoA, que se combina coa glicina formando hipurato. A glicina utilizada nesta reacción debe ser rexenerada, e así o amoníaco utilízase na reacción da glicina sintase. O fenilacetato combínase con glutamina para formar fenilacetilglutamina. Tanto o hipurato coma a fenilacetilglutamina son compostos non tóxicos que se excretan na urina.
Outras terapias son máis específicas dunha determinada deficiencia encimática. A deficiencia de N-acetilglutamato sintetase ten como resultado a ausencia do activador normal da carbamoíl fosfato sintetase I. Este estado pode tratarse administrando carbamil glutamato, un análogo do N-acetilglutamato que é efectivo como activador da carbamil fosfato sintetase I. Para as deficiencias en ornitina transcarbamilase, arxininosuccinato sintetase e arxininosuccinato liase, é útil sumplementar a dieta con arxinina. Moitos destes tratamentos deben ir acompañados dun control estrito da dieta e de suplementos dos aminoácidos esenciais. Nos poucos casos de deficiencia de arxinase, a arxinina, o substrato do encima defectivo, debe excluírse da dieta.
Anomalias do ciclo da urea que causan trastornos no ciclo da urea:
Como o fumarato producido na reacción da arxininosuccinato liase é tamén un intermediario do ciclo do ácido cítrico, os ciclos están, en principio, interconectados, proceso coñecido como o "dobre ciclo de Krebs". Porén, cada ciclo pode funcionar de xeito independente e a comunicación entre eles depende do transporte de intermediarios chave entre a mitocondria e o citosol. Varios encimas do ciclo do ácido cítrico, como a fumarase (fumarato hidratase) e a malato deshidroxenase, tamén están presentes como isoencimas no citosol. O fumarato xerado na síntese citosólica de arxinina pode, xa que logo, converterse en malato e despois en oxalacetato no citosol, e estes intermediarios poden seguir metabolizándose no citosol ou ser transportados ás mitocondrias para a súa utilización no ciclo do ácido cítrico.
O aspartato formado nas mitocondrias por transaminación entre o oxalacetato e o glutamato pode ser transportado ao citosol, onde actúa como doante de nitróxeno na reacción do ciclo da urea catalizada pola arxininosuccinato sintetase. Estas reaccións, que constitúen a desviación do aspartato-arxininosuccinato, proporcionan vínculos metabólicos entre as rutas separadas polas que se procesan os grupos amino e os esqueletos carbonados dos aminoácidos.