Plaqueta
Las '''plaquetas''' o '''trombocitos''' son pequeños fragmentos citoplasmáticos, irregulares, carentes de núcleo, de 2-3 µm de diámetro,[1] derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 11 días. Las plaquetas desempeñan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
| Plaquetas | ||
|---|---|---|
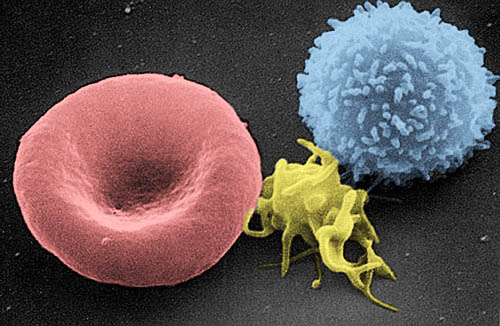 Imagen tomada con un microscopio electrónico de barrido en la que se observa, de izquierda a derecha: un glóbulo rojo, una plaqueta y un glóbulo blanco. | ||
 Representación 3D de varias plaquetas, algunas activadas (activated platelets). | ||
| Nombre y clasificación | ||
| Sinónimos |
| |
| Latín | Thrombocytus | |
| TH | H2.00.04.1.03001 | |
| Información anatómica | ||
| Sistema | Circulatorio | |
| Precursor | Megacariocito | |
|
| ||

Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebrovascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,[2] la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular. Estos dos factores de crecimiento han demostrado desempeñar un papel significativo en la regeneración y reparación del tejido conectivo.
Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.[3][4][5][6][7][8][9]
Descubrimiento
editarEntre los primeros trabajos reconocidos que describieron las plaquetas se encuentran los del biólogo y médico francés Alfred François Donné (1801-1878), el anatomista inglés George Gulliver (1804-1882), y el médico inglés William Addison (1802-1881).[10] Max Schultze (1825-1874), un anatomista alemán, fue uno de los primeros en publicar una descripción de las plaquetas como parte de un estudio de glóbulos blancos en 1865.[11] Sin embargo, los glóbulos rojos, o eritrocitos, ya eran conocidos desde van Leeuwenhoek, Schultze fue primero en publicar una descripción de las plaquetas.[12] Él describió "esférulas" mucho más pequeñas que los eritrocitos que ocasionalmente se agrupaban y participaban en colecciones de fibrina, recomendando estudios adicionales sobre estos hallazgos.
Giulio Bizzozero (1846-1901), aportó sobre los hallazgos de Schultze, usando "circulación en vivo" para estudiar las células sanguíneas de anfibios microscópicamente. Él notó especialmente que las plaquetas se agrupaban en el sitio de lesión vascular, un proceso que precedía a la formación de un coágulo. Esta observación confirmó el papel de las plaquetas en la coagulación.[13]
Cinética
editar- Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos.
- El rango fisiológico de las plaquetas es de 150-400 x 109/litro.
- Un adulto sano produce cada día alrededor de 1 x 1011 plaquetas de media.
- La expectativa de vida de las plaquetas circulantes es de 8 a 12 días.
- La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida habitualmente por el hígado y los riñones.
- Cada megacariocito produce entre 5000 y 10 000 plaquetas.
- Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado.
- Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos
editarLa función plaquetaria consiste en el mantenimiento del sistema circulatorio; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación
editarLa superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW), un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Cambio de forma
editarLas plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
Secreción de gránulos
editarLas plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos:
- Gránulos densos (contienen ADP o ATP, calcio, y serotonina).
- Gránulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y VIII).
Síntesis de tromboxano A2
editarLa activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico.
Adhesión y agregación
editarLa agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glucoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, vía glucoproteína (GPIa/IIa), al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibido por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroides anabólicos.
Reparación de heridas
editarEl coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastos del tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Evaluación de la función plaquetaria con agregometría
La agregometría plaquetaria fue desarrollada desde 1962 por Born y actualmente se considera el estándar de oro para el estudio de la función plaquetaria. Esta se realiza en un tubo de vidrio en agitación con plasma rico en plaquetas (PRP) para obligar a las plaquetas a entrar en contacto unas con otras. La adición de un agonista (ADP, adrenalina, colágeno, ácido araquidónico y ristocetina) el PRP activa a las plaquetas que se agregan en presencia de fibrinógeno; la agregación es medida con el principio turbidimetrico de Born que se basa en la diferencia en la transmisión de la luz en el PRP comparándolo con el plasma pobre en plaquetas (PPP). Debe sospecharse enfermedad cuando hay respuestas anormales con dos agonistas (excepto Adrenalina y/o ADP con los que hasta en un 15 % de los individuos sanos puede no tener respuesta) o bien, falta de respuesta al agonista ADP en dos concentraciones diferentes.
Otras funciones
editar- Retracción del coágulo
- Pro-coagulación
- Inflamación
- Señalización citoquínica
- Fagocitosis[14]
Señalización citoquínica
editarAdicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción de citoquinas, quimiosinas, y otros mediadores de la inflamación[15][16][17][18] las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
Papel en enfermedades
editarRecuentos altos y bajos
editarEl recuento de plaquetas de un individuo sano se encuentra entre 150 000 y 450 000 por μl (microlitro) de sangre (150-450 x 109/L).[19] El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad esté presente es más alta.
Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo, existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina. En la trombocitosis se puede producir trombosis, sin embargo, esto sucede principalmente cuando el recuento elevado es debido a desórdenes mieloproliferativos.
Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión está contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula la coagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L.
El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de la ciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurará hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos siete días. El ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas,[20] y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta.[21] La uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
Medicamentos
editarAgentes orales usados a menudo para alterar/suprimir la función plaquetaria:
Agentes intravenosos usados a menudo para alterar/suprimir la función plaquetaria:
Enfermedades
editarDesórdenes que provocan un recuento bajo de plaquetas:
- Trombocitopenia
- Púrpura trombocitopénica idiopática
- Púrpura trombocitopénica trombótica
- Púrpura trombocitopenica inducida por medicamentos
- Enfermedad de Gaucher
- Anemia aplásica
Trastornos Aloimunes
- Trombocitopenia fetomaterna autoinmune
- Algunas reacciones trasfusionales
Desórdenes que provocan disfunción o recuento reducido:
- Síndrome HELLP
- Síndrome urémico hemolítico
- Quimioterapia
- Dengue
- Deficiencia del almacenamiento en gránulos Alfa-Delta; es un desorden hemorrágico hereditario.[22]
Desórdenes caracterizados por recuentos elevados:
- Trombocitosis, incluyendo trombocitosis esencial (recuento elevado, ya sea reactivo o como una expresión de trastorno mieloproliferativo); puede mostrar plaquetas disfuncionales.
Desórdenes de la agregación y adherencia plaquetarios:
- Síndrome de Bernard-Soulier
- Tromboastenia de Glanzmann
- Síndrome de Scott's
- Enfermedad de von Willebrand
- Síndrome de Hermansky-Pudlak
- Síndrome de plaquetas grises
Desórdenes del metabolismo de plaquetas
- Actividad disminuida de la oxigenación, inducida o congénita
- Defectos del almacenamiento, adquirido o congénito
Desórdenes que comprometen la función plaquetaria:
Desórdenes en los cuales las plaquetas desempeñan un papel clave:
Un estudio publicado en 2021 en la revista Nature Communications demostró la existencia de alelos responsables del desarrollo de ateroesclerosis en plaquetas de pacientes afectados por la enfermedad. Se utilizaron técnicas de secuenciación genómica (WGS), eQTL para la co-localización de los loci identificados, estudios epigenómicos y consulta en bases de datos para corroborar la implicación de las variantes. Se identificaron polimorfismos para los genes PEAR1 y RGS18. EL primero únicamente tenía un SNP, mientras que para el segundo se observaron un gran número de variantes relacionadas con la enfermedad.
Respecto al alelo menor de PEAR1, se encontró evidencia de su relación con un incremento en la posibilidad de desarrollar hemorragias gastrointestinales de hasta 6 veces por encima de lo normal, debido a un descenso en el fenómeno de agregación plaquetaria inducida por una menor expresión del gen.
Para el gen RGS18, además de lo anterior, se produjeron ratones knockout para este gen de forma que se pudo observar in vivo el efecto de la supresión de este. Esto resultó en una exagerada reacción plaquetaria en respuesta a sustancias agonistas: ADP y epinefrina, además de un incremento en el taponamiento de arterias debido a la pérdida de inhibición de los receptores de la proteína G en las plaquetas del ratón. Para el alelo menor de este gen se encontraron datos que lo asociaban a mayor riesgo de trombosis tanto en pacientes de procedencia europea como afroamericana. Además, se localizaron otras 2 variantes que provocaban un descenso en la expresión de RGS18 debido a la interrupción en los sitios de unión de GATA1 y NFE2.
- Enfermedad coronaria arterial, e infarto del miocardio
- Accidente cerebrovascular
- Enfermedad arterial oclusiva periférica
- Cáncer[23]
Plasma rico en plaquetas (PRP)
editarLa preparación del plasma rico en plaquetas (PRP) de procedencia autóloga, requiere la extracción y recolección de sangre periférica del paciente, la separación de las plaquetas y el plasma de los otros elementos formes sanguíneos, y la posterior polimerización de la fibrina de dicho plasma para concentrar las plaquetas formando un gel rico en plaquetas con suficiente estabilidad como para ser implantado quirúrgicamente. Actualmente, algunos métodos comerciales para la preparación del PRP utilizan calcio y trombina bovina[8][9] o bien, trombina preparada de forma autóloga para crear una matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix). La preparación de trombina autóloga requiere tiempo y pasos adicionales, así como un mayor volumen de sangre; por otro lado, el uso de trombina bovina ha sido asociado con el desarrollo de anticuerpos contra los factores de coagulación V y XI, y la misma trombina, aumentando de esta forma el riesgo de anormalidades en la coagulación.[24][25][26] Adicionalmente, para asegurar una desgranulación de las plaquetas y la formación de un coágulo estable, se utilizan grandes cantidades de trombina, esto puede causar una liberación inmediata de los factores de crecimiento.[9]
La liberación de factores de crecimiento es desencadenada por la activación de las plaquetas, esta puede ser iniciada por una gran variedad de sustancias o estímulos como la trombina, el cloruro de calcio, el colágeno o el adenosina 5c-difosfato.[27][28]
Un coágulo sanguíneo de PRP contiene aproximadamente un 4 % de glóbulos rojos, 95 % de plaquetas y 1 % de glóbulos blancos. Las propiedades del PRP están basadas en los múltiples factores de crecimiento y de diferenciación producidos y liberados a raíz de la activación de las plaquetas. Estos factores son críticos para la regulación y estimulación del proceso curativo de lesiones. Por otro lado, existe una segunda generación del concentrado de plaquetas que recibe el nombre de matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix), la cual es un mejoramiento del PRP preparado tradicionalmente.[27]
Existe un método actual que evita el uso de trombina como activador.[29][30][31] Este sistema utiliza únicamente calcio y centrifugación para activar la polimerización de la fibrina y formar así el PRFM. PRFM, en forma de gel o una membrana densa y flexible, puede ser aplicada al paciente y la liberación de los factores de crecimiento es desencadenada por los activadores autólogos presentes en el sitio de aplicación. Este método permite una liberación gradual de los factores de crecimiento en el sitio de aplicación, que pueden emitir señales a diferentes tipos celulares para que emitan una respuesta en momentos apropiados. Estudios in vitro indican que el PRFM presenta una liberación gradual y estable de los factores de crecimiento a lo largo de 7 días.[32]
El plasma rico en plaquetas (PRP) puede obtenerse por medio de diferentes técnicas ya sean separadores celulares de propósitos generales o bien, separadores celulares para la concentración de plaquetas.[27] Muchos productos comerciales se encuentran disponibles en este campo, la mayoría de ellos obtienen resultados similares, cuyas diferencias se deben fundamentalmente al precio, tiempo, espacio requerido y la tecnología necesaria para fabricarlo. Son pocos los productos comerciales disponibles para la obtención de una matriz rica en plaquetas y fibrina como producto final.
Referencias
editar- ↑ Campbell, Neil A. (2008). Biology (8th edición). Londres: Pearson Education. p. 912. ISBN 978-0-321-53616-7. «Platelets are pinched-off cytoplasmic fragments of specialized bone marrow cells. They are about 2-3 µm in diameter and have no nuclei. Platelets serve both structural and molecular functions in blood clotting.»
- ↑ Matón, Anthea; Jean Hopkins, Charles William McLaughlin, Susan Johnson, Maryanna Quon Warner, David LaHart, Jill D. Wright (1993). Human Biology and Health. Englewood Cliffs, New Jersey, USA: Prentice Hall. ISBN 0-13-981176-1.
- ↑ O’Connell S, Impeduglia T, Hessler K, Wang XJ, Carroll R, Dardik H. Autologous platelet-rich fibrin matrix as cell therapy in the healing of chronic lower-extremity ulcers. Wound Rep Reg 2008; 16:749-756.
- ↑ Sánchez M, Anitua E, Azofra J, Andía I, Padilla S, Mujika I. Comparison of surgically repaired Achilles tendon tears using platelet-rich fibrin matrices. The American Journal of Sports Medicine 2007; 35 (2): 245-51.
- ↑ Knighton DR, Ciresi KF, Fiegel VD, Austin LL, Butler ELL. Classification and treatment of chronic nonhealing wounds: successful treatment with autologous platelet-derived wound healing factors (PDWHF). Ann surg 1986; 204:322-30.
- ↑ Knighton DR, Ciresi K, Fiegel VD, Schumerth S, Butler E, Cerra F. Stimulation of repair in chronic, non healing, cutaneous ulcers using platelet-derived wound healing formula. Surg Gynecol Obstet 1990; 170:56-60.
- ↑ Celotti F, Colciago A, Negri-Cesi P, Pravettoni A, Zaninetti R, Sacchi MC. Effect of platelet-rich plasma on migration and proliferation of SaOS-2 osteoblasts: role of platelet-derived growth factor and transforming growth factor-β. Wound Rep Regen 2006; 14:195-202.
- ↑ a b McAleer JP, Sharma S, Kaplan EM, Perisch G. Use of autologous platelet concentrate in a nonhealing lower extremity wound. Adv Skin Wound Care 2006; 19:354-63.
- ↑ a b c Driver VR, Hanft J, Fylling CP, Beriou JM. Arospective, randomized, controlled trial of autologous platelet-rich plasma gel for the treatment of diabetic foot ulcers. Ostomy Wound Manage 2006;52:68-87.
- ↑ Izaguirre-Avila, Raúl (1997). «El descubrimiento de las plaquetas.». Revista Biomédica 8 (3): 196-208.
- ↑ Brewer DB. Max Schultze (1864), G. Bizzozero (1882) and the discovery of the platelet. Br J Haematol 2006;133:251-8. PMID 16643426.
- ↑ Schultze M. Ein heizbarer Objecttisch und seine Verwendung bei Untersuchungen des Blutes. Arch Mikrosc Anat 1865;1:1-42.
- ↑ Bizzozero J. Über einen neuen Forrnbestandteil des Blutes und dessen Rolle bei der Thrombose und Blutgerinnung. Arch Pathol Anat Phys Klin Med 1882;90:261-332.
- ↑ Movat H.Zet al. (1965). «Platelet Phagocytosis and Aggregation». Journal of Cell Biology 27: 531-543. PMID 4957257. doi:10.1083/jcb.27.3.531.
- ↑ Weyrich A.S. et al. (2004). «Platelets: signaling cells inside the immune continuum.». Trends Immunol 25: 489-495.
- ↑ Wagner D.D. et al. (2003). «Platelets in inflammation and thrombosis.». Thromb Vasc Biol 23: 2131-2137.
- ↑ Diacovo T.G. et al. (1996). «Platelet-mediated lymphocyte delivery to high endothelial venules.». Science 273: 252-255.
- ↑ Iannacone M. et al. (2005). «Platelets mediate cytotoxic T lymphocyte-induced liver damage». Nat Med 11: 1167-1169.
- ↑ Kumar & Clark (2005). «8». Clinical Medicine (Sixth edición). Elsevier Saunders. pp. 469. ISBN 0702027634. Consultado el 26 de marzo de 2009.
- ↑ «Platelet Function after Taking Ibuprofen for 1 Week». Archivado desde el original el 25 de julio de 2008. Consultado el 26 de agosto de 2008.
- ↑ «Ibuprofen protects platelet cyclooxygenase from irreversible inhibition by aspirin». Consultado el 26 de agosto de 2008.
- ↑ Alpha-delta platelet storage pool deficiency in three generations - Platelets
- ↑ McCarty OJT. et al. (2000). «Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesión under dynamic flow conditions». Blood 96: 1789-1197.
- ↑ Mazzucco L, Medici D, Serra M, Panizza R, Rivara G, Orecchia S, Libener R, Cattna E, Levis A, Betta PG, Borzini P. «The use of autologous platelet gel to treat difficult to heal wounds: a pilot study.» Transfusion 2004; 44: 1013-8.
- ↑ Kevy SV, Jacobson MS. Comparison of methods for point of care preparation of autologous platelet gel. JECT 2004; 36: 28-35.
- ↑ Bänninger H, Hardegger T, Tobler A, Barth A, Schüpbach P, Reinhart W, Lämmle B, Furlan M. Fibrin glue in surgery: frequent development of inhibitors of bovine thrombin and human factor V. Br J Haematol 1993; 85: 528-32.
- ↑ a b c Sunitha R, Munirathnam N. Platelet-rich fibrin: Evolution of a second-generation platelet concentrate. Indian J Dent Res 2008; 19(1):42-46.
- ↑ Marx RE, Carlson ER, Eichstaedt RM, Schimmele ST, Strauss JE, Georgeff KR. «Platelet-rich plasma: Growth factor enhancement for bone grafts.» Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998;85:638-46.
- ↑ Ortel TL, Mercer MC, Thames EH, Moore KD, Lawson JH. Immunologic impact and clinical outcomes after surgical exposure to bovine thrombin. Ann Surg 2001; 233: 88-96.
- ↑ Schoenecker JG, Johnson RK, Lesher AP, Day JD, Love SD, Hoffman MR, Ortel TL, Parker W, Lawson JH. «Exposure of mice to topical bovine thrombin induces systemic autoimmunity.» Am J Pathol 2001; 159: 1957-69.
- ↑ Vercellino V, Carbone V, Griffa A. «The use of autologous materials for treatment of large maxillary cysts: filling of the residual post-cystectomy cavity.» 5th Congress, Society Odontostonatologica Italiano, Turin, Italy, December 1, 2000.
- ↑ Gosch C, Zeichner A, Carroll R, Bois J. «Evaluation of an autologous platelet rich fibrin matrix technology for diabetic foot ulcer treatment.» Wound Rep Regen 2007; 15: A38.
32. Vargas RAG, Hernández HD, Villa MR. Evaluación de la función plaquetaria con agregometría. Rev. Hemost Trombo 2010; 3(2): 29-38.
33. Keramati, A. R., Chen, M. H., Rodriguez, B. A., Yanek, L. R., Bhan, A., Gaynor, B. J., ... & Johnson, A. D. (2021). Genome sequencing unveils a regulatory landscape of platelet reactivity. Nature Communications, 12(1), 1-13.
Enlaces externos
editarWikimedia Commons alberga una categoría multimedia sobre Plaqueta.