Ring expansion and contraction
This article has multiple issues. Please help improve it or discuss these issues on the talk page. (Learn how and when to remove these messages)
|
Ring expansion and ring contraction reactions expand or contract rings, usually in organic chemistry. The term usually refers to reactions involve making and breaking C-C bonds,[1] Diverse mechanisms lead to these kinds of reactions.
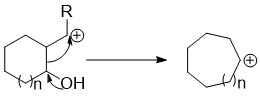
Demyanov ring contraction and expansion
[edit]These reactions entail diazotization of aminocyclobutanes and aminocyclopropanes. Loss of N2 from the diazo cations results in secondary carbocations, which tend to rearrange and then undergo hydrolysis. The reaction converts aminocyclobutane into a mixture of hydroxycyclobutane and hydroxymethylcyclopropane. These reactions produce an equilibrating mixture of two carbocations:[2]
- C4H+7 ⇌ C3H5CH+2
Carbenoid ring contractions
[edit]
In the Arndt–Eistert reaction, an α-diazoketone is induced to release N2, resulting in a highly reactive sextet carbon center adjacent to the carbonyl. Such species convert by a Wolff rearrangement to give an ester in the presence of alcohols. When applied to cyclic α-diazoketones, ring contraction occurs.[3][4] In the case of steroids, this reaction has been used to convert cyclopentanone groups to cyclobutanyl derivatives.[5]
Ring expansion reactions
[edit]
Ring expansions can allow access to larger systems that can be difficult to synthesize otherwise.[6] Rings can be expanded by attack of the ring onto an outside group already appended to the ring (a migration/insertion), opening of a bicycle to a single larger ring, or coupling a ring closing with an expansion.[1] These expansions can be further broken down by what type of atom they incorporate (a carbon or a heteroatom) into the expanded ring.
Carbon insertion through migration to an exocyclic group
[edit]These reactions have the general features of having an exocyclic leaving group on a carbon adjacent to the ring and an electron donating group on the ring capable of initiating a migration of an endocyclic bond.
A common migration introduction of carbon is a pinacol rearrangement.[1] While this reaction refers specifically to a vicinal dihydroxide rearrangement, there are other pinacol type rearrangements that proceed through the same general mechanism such as the Tiffeneau–Demjanov rearrangement. These "semipinacol rearrangements" occur under milder conditions and are thus preferable in complex syntheses.[7] These reactions are useful beyond simply expanding a ring because the exocyclic group attacked may also have other functionality appended to it besides the leaving group. The group to which the endocyclic bond migrates can also be selectively added to the ring based on the functionality already present, for example 1,2 addition into a cyclic ketone.
Carbon insertion through opening of a bicycle
[edit]
A common method for expanding a ring involves opening cyclopropane-containing bicyclic intermediate. The strategy can start with a Simmons-Smith-like cyclopropanation of a cyclic alkene.[8]
A related cyclopropane-based ring expansion is the Buchner ring expansion. The Buchner ring expansion is used to convert arenes to cycloheptatrienes. The Buchner ring expansion is encouraged to open to the desired product by placing electron withdrawing groups on the carbon added. In order to perform the ring opening on saturated bicyclic molecules the cyclopropane must be introduced such that a neighboring group can facilitate the expansion or the ring must be opened by attackate the expansion[9] or the ring must be opened by attack from an outside group.[10]
Ring opening as a means of ring expansion can also be applied to larger systems to give access to even larger ring syscyclization. The Grob fragmentation can be applied as an example of such an expansion. Like the pinacol type migration the Grob fragmentation relies on an electron donating group to promote the bond migration and encourage the leaving group to be expelled. In this case the electron donating group can be a pseudo electron donating group which is capable of eliminating and donating an electron pair into the carbon with the breaking bond. Working with two smaller rings can allow for elaboration of two parts of the molecule separately before working with the expanded ring. The Dowd-Beckwith ring expansion reaction is also capable of adding several carbons to a ring at a time, and is a useful tool for making large rings.[11] While it proceeds through an intermediate bicycle the final cyclization and ring opening take place within the same radical reaction.[12] This expansion is useful because it allows the expansion of a beta-ketoester to a large cyclic ketone which can easily be elaborated using either the cyclic ketone or the exocyclic ester.

Heteroatom insertion reactions
[edit]Heteroatom additions to rings can occur through ring expansions if not they are not done through de-novo ring synthesis.[13] These introductions are primarily ring expansions because they often take place through migration/insertion pathways similar to those mentioned above for carbon. Examples include high impact applications of the Beckmann rearrangement (for introduction of nitrogen into codeine)[14] and the Baeyer-Villiger oxidation (introduction of oxygen to cage-annulated ethers)[15] in synthesis. Both occur with the expulsion of a leaving group as the alkyl group migrates onto the exocyclic heteroatom, which is strikingly similar to the pinacol type rearrangement.
Ring contraction reactions
[edit]
Ring contractions are useful for making smaller, more strained rings from larger rings. The impetus for making these rings comes from the difficulty associated with making a fully elaborated small ring when such a ring could more easily be made from an elaborated larger ring, from which an atom can be excised, or that the original larger scaffold is more accessible.[16]
Ring contractions are easily characterized simply by the reactive intermediate which performs the contraction. The standard intermediates are anionic, cationic, and carbenoid.[17]
Favorskii rearrangement
[edit]The Favorskii rearrangement is a classic anionic ring contraction.[18] It proceeds through a carbanion that attacks an endocyclic carbon and expels a leaving group (a halide) forming a bicyclic molecule with rings smaller than the original. The bicycle is then opened by nucleophilic attack on the ketone to give the contracted product.[19] This reaction has been used to convert cyclohexanone to the methyl ester of cyclopropanecarboxylic acid.

An alternative to the standard Favorskii rearrangement, is to perform what can be thought of as a negative pinacol rearrangement where an anionic group encourages a bond aligned with a leaving group to migrate and expel the leaving group, which has been used in several syntheses.[17] It should also be noted that the so-called "quasi-Favorskii rearrangement" proceeds without an additional nucleophile to form the final contracted product.

Cation contractions
[edit]The cationic rearrangement contraction proceeds through the loss of a leaving group and the migration of an endocyclic bond to the carbocation. Pinacol type rearrangements are often used for this type of contraction.[20] Like the expansion reaction this proceeds with an electron donating group aiding in the migration.
Contraction reactions of one ring can be coupled with an expansion of another to give an unequal bicycle from equally sized fused ring. These cationic rearrangements have found use to synthesize the cores of complex molecules.[21]
Further reading
[edit]- Redmore, D.; Gutsche, C.D. Carbocyclic Ring Expansion Reactions, Academic Press, NY, 1968,
References
[edit]- ^ a b c Kantorowski, E.J.; Kurth, M.J. (2000). "Expansion to seven-membered rings" (PDF). Tetrahedron. 56 (26): 4317–4353. doi:10.1016/S0040-4020(00)00218-0. S2CID 34628258. Archived from the original (PDF) on August 18, 2019.
- ^ Smith, Michael B.; March, Jerry (2006). March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. pp. 1588–1592. doi:10.1002/0470084960. ISBN 9780470084960.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Smith, Michael B.; March, Jerry (2006). March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. p. 1599. doi:10.1002/0470084960. ISBN 9780470084960.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Kirmse, W. (July 2002). "100 Years of the Wolff Rearrangement". European Journal of Organic Chemistry. 2002 (14): 2193. doi:10.1002/1099-0690(200207)2002:14<2193::AID-EJOC2193>3.0.CO;2-D.
- ^ Thomas N. Wheeler and J. Meinwald (1972). "Formation and Photochemical Wolff Rearrangement of Cyclic α-Diazo Ketones: d-Norandrost-5-en-3β-ol-16-carboxylic Acids". Organic Syntheses. 52: 53. doi:10.15227/orgsyn.052.0053.
- ^ Casadei, M.A.; Calli, C.; Mandolini, L. (February 1, 1984). "Ring-closure reactions. 22. Kinetics of cyclization of diethyl (.omega.-bromoalkyl)malonates in the range of 4- to 21-membered rings. Role of ring strain". Journal of the American Chemical Society. 106 (4): 1051–1056. doi:10.1021/ja00316a039.
- ^ Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions. Elsevier. p. 350. ISBN 978-0-12-429785-2. OCLC 1107566236.
- ^ "One-Carbon Ring Expansion of Cycloalkanones to Conjugated Cycloalkenones: 2-Cyclohepten-1-One". Organic Syntheses. 59: 113. 1979. doi:10.15227/orgsyn.059.0113.
- ^ Bieräugel, H.; Akkerman, J. M.; Armande, J. C. L.; Pandit, U. K. (1974). "A specific insertion of carbenes into carbon-carbon bonds". Tetrahedron Letters. 15 (33): 2817–2820. doi:10.1016/S0040-4039(01)91751-4.
- ^ Hoberg, J.O.; Bozell, J.J. (September 1995). "Cyclopropanation and ring-expansion of unsaturated sugars". Tetrahedron Letters. 36 (38): 6831–6834. doi:10.1016/0040-4039(95)01387-W.
- ^ Hierold, J.; Lupton, D.W. (July 2012). "Synthesis of Spirocyclic γ-Lactones by Cascade Beckwith–Dowd Ring Expansion/Cyclization". Organic Letters. 14 (13): 3412–3415. doi:10.1021/ol301387t. PMID 22691029.
- ^ Dowd, P.; Choi; S. C. J. Am. Chem. Soc. 1987, 3493–3494
- ^ McMurry, John (2008). Organic Chemistry 7th Ed. pp. 945–946. ISBN 978-0-495-11258-7.
- ^ White, J. D.; Hrnciar, P.; Stappenbeck, F. (1999). "Asymmetric Total Synthesis of (+)-Codeine via Intramolecular Carbenoid Insertion". Journal of Organic Chemistry. 63 (21): 7871–7884. doi:10.1021/jo990905z.
- ^ Marchand, A. P.; Kumar, V. S.; Hariprakasha, H. K. (2001). "Synthesis of Novel Cage Oxaheterocycles". Journal of Organic Chemistry. 66 (6): 2072–2077. doi:10.1021/jo001611c. PMID 11300903.
- ^ Silva, Luiz F., Jr. (2002). "Construction of cyclopentyl units by ring contraction reactions". Tetrahedron. 58 (45): 9137–9161. doi:10.1016/S0040-4020(02)00990-0.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ a b Myers, Andrew. "Methods for Ring Contraction" (PDF). Retrieved November 30, 2014 – via Harvard University Department of Chemistry and Chemical Biology.
- ^ Chenier, Philip J. (1978). "The Favorskii Rearrangement in Bridged Polycyclic Compounds". Journal of Chemical Education. 55 (5): 286–291. Bibcode:1978JChEd..55..286C. doi:10.1021/ed055p286.
- ^ Smith, Michael B.; March, Jerry (2006). March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. pp. 1595–1596. doi:10.1002/0470084960. ISBN 9780470084960.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Song, Zhen-Lei; Fan, Chun-An; Tu, Yong-Qiang (2011). "Semipinacol Rearrangement in Natural Product Synthesis". Chemical Reviews. 111 (11): 7523–7556. doi:10.1021/cr200055g. PMID 21851053.
- ^ Büchi, G.; Hofheinz, W.; Paukstelis, J. V. (November 1969). "Synthesis of (-)-aromadendrene and related sesquiterpenes". Journal of the American Chemical Society. 91 (23): 6473–6478. doi:10.1021/ja01051a051.