Pharmacology of ethanol



The pharmacology of ethanol involves both pharmacodynamics (how it affects the body) and pharmacokinetics (how the body processes it). In the body, ethanol primarily affects the central nervous system, acting as a depressant and causing sedation, relaxation, and decreased anxiety. The complete list of mechanisms remains an area of research, but ethanol has been shown to affect ligand-gated ion channels, particularly the GABAA receptor.
After oral ingestion, ethanol is absorbed via the stomach and intestines into the bloodstream. Ethanol is highly water-soluble and diffuses passively throughout the entire body, including the brain. Soon after ingestion, it begins to be metabolized, 90% or more by the liver. One standard drink is sufficient to almost completely saturate the liver's capacity to metabolize alcohol.[citation needed] The main metabolite is acetaldehyde, a toxic carcinogen. Acetaldehyde is then further metabolized into ionic acetate by the enzyme aldehyde dehydrogenase (ALDH). Acetate is not carcinogenic and has low toxicity,[9] but has been implicated in causing hangovers.[10][11] Acetate is further broken down into carbon dioxide and water and eventually eliminated from the body through urine and breath. 5 to 10% of ethanol is excreted unchanged in the breath, urine, and sweat.
History
[edit]
Beginning with the Gin Craze, excessive drinking and drunkenness developed into a major problem for public health.[12][13] In 1874, Francis E. Anstie's experiments showed that the amounts of alcohol eliminated unchanged in breath, urine, sweat, and feces were negligible compared to the amount ingested, suggesting it was oxidized within the body.[14] In 1902, Atwater and Benedict estimated that alcohol yielded 7.1 kcal of energy per gram consumed and 98% was metabolized.[15] In 1922, Widmark published his method for analyzing the alcohol content of fingertip samples of blood.[16] Through the 1930s, Widmark conducted numerous studies and formulated the basic principles of ethanol pharmacokinetics for forensic purposes,[17] including the eponymous Widmark equation. In 1980, Watson et al. proposed updated equations based on total body water instead of body weight.[18] The TBW equations have been found to be significantly more accurate due to rising levels of obesity worldwide.[19]
Pharmacodynamics
[edit]The principal mechanism of action for ethanol has proven elusive and remains not fully understood.[20][21] Identifying molecular targets for ethanol is unusually difficult, in large part due to its unique biochemical properties.[21] Specifically, ethanol is a very low molecular weight compound and is of exceptionally low potency in its actions, causing effects only at very high (millimolar mM) concentrations.[21][22] For these reasons, it is not possible to employ traditional biochemical techniques to directly assess the binding of ethanol to receptors or ion channels.[21][22] Instead, researchers have had to rely on functional studies to elucidate the actions of ethanol.[21] Even at present, no binding sites have been unambiguously identified and established for ethanol. Studies have published strong evidence for certain functions of ethanol in specific systems, but other laboratories have found that these findings do not replicate with different neuronal types and heterologously expressed receptors.[23] Thus, there remains lingering doubt about the mechanisms of ethanol listed here, even for the GABAA receptor, the most-studied mechanism.[24]
In the past, alcohol was believed to be a non-specific pharmacological agent affecting many neurotransmitter systems in the brain,[25] but progress has been made over the last few decades.[26][21] It appears that it affects ion channels, in particular ligand-gated ion channels, to mediate its effects in the CNS.[20][26][27][21] In some systems, these effects are facilitatory, and in others inhibitory. Moreover, although it has been established that ethanol modulates ion channels to mediate its effects,[27] ion channels are complex proteins, and their interactions and functions are complicated by diverse subunit compositions and regulation by conserved cellular signals (e.g. signaling lipids).[20][21]
Alcohol is also converted into phosphatidylethanol (PEth, an unnatural lipid metabolite) by phospholipase D2. This metabolite competes with PIP2 agonist sites on lipid-gated ion channels.[28][29] The result of these direct effects is a wave of further indirect effects involving a variety of other neurotransmitter and neuropeptide systems.[25] This presents a novel indirect mechanism and suggests that a metabolite, not the ethanol itself, could cause the behavioural or symptomatic effects of alcohol intoxication. Many of the primary targets of ethanol are known to bind PIP2 including GABAA receptors,[30] but the role of PEth needs to be investigated further.
List of known actions in the central nervous system
[edit]Ethanol has been reported to possess the following actions in functional assays at varying concentrations:[22]
- GABAA receptor: positive allosteric modulator (primarily of δ subunit-containing receptors)[31][32]
- NMDA receptor: negative allosteric modulator[33][31][34]
- AMPA receptor: negative allosteric modulator[33]
- Kainate receptor: negative allosteric modulator[33]
- Glycine receptor: positive allosteric modulator[35]
- Serotonin 5-HT3 receptor: positive allosteric modulator[35][34]
- Opioid receptor: endogenous positive allosteric modulator[33]
- Muscarinic acetylcholine receptor: positive allosteric modulator.
- Nicotinic acetylcholine receptor: positive allosteric modulator[36][37][38]
- Glycine reuptake inhibitor[39]
- Adenosine reuptake inhibitor[40]
- L-type calcium channel: channel blocker
- GIRK: channel opener
- Voltage-gated calcium channel[20][26][27]
- Dihydropyridine-sensitive L-type Ca2+ channels[41]
- BK channel modulation[42]
- G-protein-activated inwardly rectifying K+ channels[43]
- Brain medulla: Decreased levels of nitric oxide[44]
- Mesolimbic pathway: Increased levels of dopamine and endogenous opioids, secondary to other actions[45][31]
Many of these actions have been found to occur only at very high concentrations that may not be pharmacologically significant at recreational doses of ethanol, and it is unclear how or to what extent each of the individual actions is involved in the effects of ethanol.[21] Some of the actions of ethanol on ligand-gated ion channels, specifically the nicotinic acetylcholine receptors and the glycine receptor, are dose-dependent, with potentiation or inhibition occurring dependent on ethanol concentration.[22] This seems to be because the effects of ethanol on these channels are a summation of positive and negative allosteric modulatory actions.[22]
GABAA receptors
[edit]
Ethanol has been found to enhance GABAA receptor-mediated currents in functional assays.[20][21] Ethanol has long shown a similarity in its effects to positive allosteric modulators of the GABAA receptor like benzodiazepines, barbiturates, and various general anesthetics.[20][21] Some of these effects include anxiolytic, anticonvulsant, sedative, and hypnotic effects, cognitive impairment, and motor incoordination.[46] In accordance, it was theorized and widely believed that the primary mechanism of action of ethanol is GABAA receptor positive allosteric modulation.[20][21] However, other ion channels are involved in its effects as well.[26][21] Although ethanol exhibits positive allosteric binding properties to GABAA receptors, its effects are limited to pentamers containing the δ-subunit rather than the γ-subunit.[21] Ethanol potentiates extrasynaptic δ subunit-containing GABAA receptors at behaviorally relevant (as low as 3 mM) concentrations,[20][21][47] but γ subunit receptors are enhanced only at far higher concentrations (> 100 mM) that are in excess of recreational concentrations (up to 50 mM).[20][21][48]
GABAA receptors containing the δ-subunit have been shown to be located exterior to the synapse and are involved with tonic inhibition rather than its γ-subunit counterpart, which is involved in phasic inhibition.[46] The δ-subunit has been shown to be able to form the allosteric binding site which makes GABAA receptors containing the δ-subunit more sensitive to ethanol concentrations, even to moderate social ethanol consumption levels (30mM).[49] While it has been shown by Santhakumar et al. that GABAA receptors containing the δ-subunit are sensitive to ethanol modulation, depending on subunit combinations receptors could be more or less sensitive to ethanol.[50] It has been shown that GABAA receptors that contain both δ and β3-subunits display increased sensitivity to ethanol.[21] One such receptor that exhibits ethanol insensitivity is α3-β6-δ GABAA.[50] It has also been shown that subunit combination is not the only thing that contributes to ethanol sensitivity. Location of GABAA receptors within the synapse may also contribute to ethanol sensitivity.[46]
Ro15-4513, a close analogue of the benzodiazepine antagonist flumazenil (Ro15-1788), has been found to bind to the same site as ethanol and to competitively displace it in a saturable manner.[21][47] In addition, Ro15-4513 blocked the enhancement of δ subunit-containing GABAA receptor currents by ethanol in vitro.[21] In accordance, the drug has been found to reverse many of the behavioral effects of low-to-moderate doses of ethanol in rodents, including its effects on anxiety, memory, motor behavior, and self-administration.[21][47] Taken together, these findings suggest a binding site for ethanol on subpopulations of the GABAA receptor with specific subunit compositions via which it interacts with and potentiates the receptor.[20][21][47][51]
Calcium channel blocking
[edit]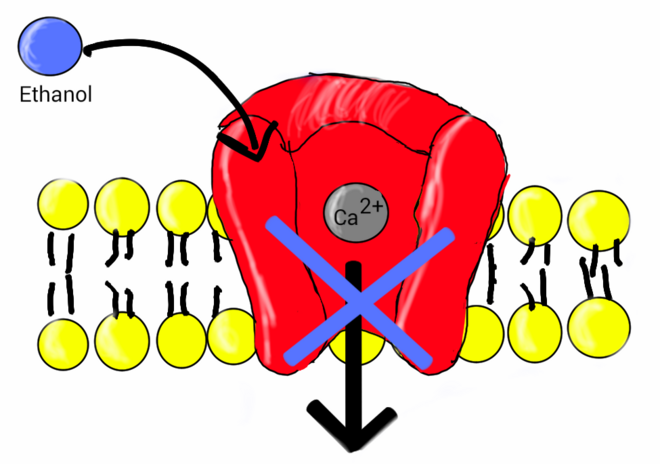
Research indicates ethanol is involved in the inhibition of L-type calcium channels. One study showed the nature of ethanol binding to L-type calcium channels is according to first-order kinetics with a Hill coefficient around 1. This indicates ethanol binds independently to the channel, expressing noncooperative binding.[41] Early studies showed a link between calcium and the release of vasopressin by the secondary messenger system.[52] Vasopressin levels are reduced after the ingestion of alcohol.[53] The lower levels of vasopressin from the consumption of alcohol have been linked to ethanol acting as an antagonist to voltage-gated calcium channels (VGCCs). Studies conducted by Treistman et al. in the aplysia confirm inhibition of VGCC by ethanol. Voltage clamp recordings have been done on the aplysia neuron. VGCCs were isolated and calcium current was recorded using patch clamp technique having ethanol as a treatment. Recordings were replicated at varying concentrations (0, 10, 25, 50, and 100 mM) at a voltage clamp of +30 mV. Results showed calcium current decreased as concentration of ethanol increased.[54] Similar results have shown to be true in single-channel recordings from isolated nerve terminal of rats that ethanol does in fact block VGCCs.[55]
Studies done by Katsura et al. in 2006 on mouse cerebral cortical neurons, show the effects of prolonged ethanol exposure. Neurons were exposed to sustained ethanol concentrations of 50 mM for 3 days in vitro. Western blot and protein analysis were conducted to determine the relative amounts of VGCC subunit expression. α1C, α1D, and α2/δ1 subunits showed an increase of expression after sustained ethanol exposure. However, the β4 subunit showed a decrease. Furthermore, α1A, α1B, and α1F subunits did not alter in their relative expression. Thus, sustained ethanol exposure may participate in the development of ethanol dependence in neurons.[56]
Other experiments done by Malysz et al. have looked into ethanol effects on voltage-gated calcium channels on detrusor smooth muscle cells in guinea pigs. Perforated patch clamp technique was used having intracellular fluid inside the pipette and extracellular fluid in the bath with added 0.3% vol/vol (about 50-mM) ethanol. Ethanol decreased the Ca2+
current in DSM cells and induced muscle relaxation. Ethanol inhibits VGCCs and is involved in alcohol-induced relaxation of the urinary bladder.[57]
Rewarding and reinforcing actions
[edit]
The reinforcing effects of alcohol consumption are mediated by acetaldehyde generated by catalase and other oxidizing enzymes such as cytochrome P-4502E1 in the brain.[60] Although acetaldehyde has been associated with some of the adverse and toxic effects of ethanol, it appears to play a central role in the activation of the mesolimbic dopamine system.[45]
Ethanol's rewarding and reinforcing (i.e., addictive) properties are mediated through its effects on dopamine neurons in the mesolimbic reward pathway, which connects the ventral tegmental area to the nucleus accumbens (NAcc).[61][62] One of ethanol's primary effects is the allosteric inhibition of NMDA receptors and facilitation of GABAA receptors (e.g., enhanced GABAA receptor-mediated chloride flux through allosteric regulation of the receptor).[31] At high doses, ethanol inhibits most ligand-gated ion channels and voltage-gated ion channels in neurons as well.[31]
With acute alcohol consumption, dopamine is released in the synapses of the mesolimbic pathway, in turn heightening activation of postsynaptic D1 receptors.[61][62] The activation of these receptors triggers postsynaptic internal signaling events through protein kinase A, which ultimately phosphorylate cAMP response element binding protein (CREB), inducing CREB-mediated changes in gene expression.[61][62]
With chronic alcohol intake, consumption of ethanol similarly induces CREB phosphorylation through the D1 receptor pathway, but it also alters NMDA receptor function through phosphorylation mechanisms;[61][62] an adaptive downregulation of the D1 receptor pathway and CREB function occurs as well.[61][62] Chronic consumption is also associated with an effect on CREB phosphorylation and function via postsynaptic NMDA receptor signaling cascades through a MAPK/ERK pathway and CAMK-mediated pathway.[62] These modifications to CREB function in the mesolimbic pathway induce expression (i.e., increase gene expression) of ΔFosB in the NAcc,[62] where ΔFosB is the "master control protein" that, when overexpressed in the NAcc, is necessary and sufficient for the development and maintenance of an addictive state (i.e., its overexpression in the nucleus accumbens produces and then directly modulates compulsive alcohol consumption).[62][63][64][65]
Relationship between concentrations and effects
[edit]| mg/dL | mM | % v/v | Effects |
|---|---|---|---|
| 50 | 11 | 0.05% | Euphoria, talkativeness, relaxation, happiness, gladness, pleasure, joyfulness. |
| 100 | 22 | 0.1% | Central nervous system depression, anxiety suppression, stress suppression, sedation, nausea, possible vomiting. Impaired motor, memory, cognition and sensory function. |
| >140 | 30 | >0.14% | Decreased blood flow to brain, slurred speech, double or blurry vision. |
| 300 | 65 | 0.3% | Stupefaction, confusion, numbness, dizziness, loss of consciousness. |
| 400 | 87 | 0.4% | Ethylic intoxication, drunkenness, inebriation, alcohol poisoning or possible death. |
| 500 | 109 | >0.55% | Unconsciousness, coma and death. |
Recreational concentrations of ethanol are typically in the range of 1 to 50 mM.[48][20] Very low concentrations of 1 to 2 mM ethanol produce zero or undetectable effects except in alcohol-naive individuals.[48] Slightly higher levels of 5 to 10 mM, which are associated with light social drinking, produce measurable effects including changes in visual acuity, decreased anxiety, and modest behavioral disinhibition.[48] Further higher levels of 15 to 20 mM result in a degree of sedation and motor incoordination that is contraindicated with the operation of motor vehicles.[48] In jurisdictions in the U.S., maximum blood alcohol levels for legal driving are about 17 to 22 mM.[67][68] In the upper range of recreational ethanol concentrations of 20 to 50 mM, depression of the central nervous system is more marked, with effects including complete drunkenness, profound sedation, amnesia, emesis, hypnosis, and eventually unconsciousness.[48][67] Levels of ethanol above 50 mM are not typically experienced by normal individuals and hence are not usually physiologically relevant; however, such levels – ranging from 50 to 100 mM – may be experienced by alcoholics with high tolerance to ethanol.[48] Concentrations above this range, specifically in the range of 100 to 200 mM, would cause death in all people except alcoholics.[48]
As drinking increases, people become sleepy or fall into a stupor. After a very high level of consumption[vague], the respiratory system becomes depressed and the person will stop breathing. Comatose patients may aspirate their vomit (resulting in vomitus in the lungs, which may cause "drowning" and later pneumonia if survived). CNS depression and impaired motor coordination along with poor judgment increase the likelihood of accidental injury occurring. It is estimated that about one-third of alcohol-related deaths are due to accidents and another 14% are from intentional injury.[69]
In addition to respiratory failure and accidents caused by its effects on the central nervous system, alcohol causes significant metabolic derangements. Hypoglycaemia occurs due to ethanol's inhibition of gluconeogenesis, especially in children, and may cause lactic acidosis, ketoacidosis, and acute kidney injury. Metabolic acidosis is compounded by respiratory failure. Patients may also present with hypothermia.
Pharmacokinetics
[edit]The pharmacokinetics of ethanol are well characterized by the ADME acronym (absorption, distribution, metabolism, excretion). Besides the dose ingested, factors such as the person's total body water, speed of drinking, the drink's nutritional content, and the contents of the stomach all influence the profile of blood alcohol content (BAC) over time. Breath alcohol content (BrAC) and BAC have similar profile shapes, so most forensic pharmacokinetic calculations can be done with either. Relatively few studies directly compare BrAC and BAC within subjects and characterize the difference in pharmacokinetic parameters. Comparing arterial and venous BAC, arterial BAC is higher during the absorption phase and lower in the postabsorptive declining phase.[13]
Endogenous production
[edit]
All organisms produce alcohol in small amounts by several pathways, primarily through fatty acid synthesis,[70] glycerolipid metabolism,[71] and bile acid biosynthesis pathways.[72] Fermentation is a biochemical process during which yeast and certain bacteria convert sugars to ethanol, carbon dioxide, as well as other metabolic byproducts.[73][74] The average human digestive system produces approximately 3 g of ethanol per day through fermentation of its contents.[75] Such production generally does not have any forensic significance because the ethanol is broken down before significant intoxication ensues. These trace amounts of alcohol range from 0.1 to 0.3 μg/mL in the blood of healthy humans, with some measurements as high as 1.6 μg/mL (0.002 g/L).[76]
Auto-brewery syndrome is a condition characterized by significant fermentation of ingested carbohydrates within the body. In rare cases, intoxicating quantities of ethanol may be produced, especially after eating meals. Claims of endogenous fermentation have been attempted as a defense against drunk driving charges, some of which have been successful, but the condition is under-researched.[77]
Absorption
[edit]
Ethanol is most commonly ingested by mouth,[2] but other routes of administration are possible, such as inhalation, enema, or by intravenous injection.[4][78] With oral administration, the ethanol is absorbed into the portal venous blood through the mucosa of the gastrointestinal tract, such as in the oral cavity, stomach, duodenum, and jejunum.[13] The oral bioavailability of ethanol is quite high, with estimates ranging from 80% at a minimum[2][3] to 94%-96%.[79] The ethanol molecule is small and uncharged, and easily crosses biological membranes by passive diffusion.[80] The absorption rate of ethanol is typically modeled as a first-order kinetic process depending on the concentration gradient and specific membrane. The rate of absorption is fastest in the duodenum and jejunum, owing to the larger absorption surface area provided by the villi and microvilli of the small intestines. Gastric emptying is therefore an important consideration when estimating the overall rate of absorption in most scenarios;[13] the presence of a meal in the stomach delays gastric emptying,[4][78] and absorption of ethanol into the blood is consequently slower.[81] Due to irregular gastric emptying patterns, the rate of absorption of ethanol is unpredictable, varying significantly even between drinking occasions.[13] In experiments, aqueous ethanol solutions have been given intravenously or rectally to avoid this variation.[13] The delay in ethanol absorption caused by food is similar regardless of whether food is consumed just before, at the same time, or just after ingestion of ethanol.[4] The type of food, whether fat, carbohydrates, or protein, also is of little importance.[78] Not only does food slow the absorption of ethanol, but it also reduces the bioavailability of ethanol, resulting in lower circulating concentrations.[4]
Regarding inhalation, early experiments with animals showed that it was possible to produce significant BAC levels comparable to those obtained by injection, by forcing the animal to breathe alcohol vapor.[82] In humans, concentrations of ethanol in air above 10 mg/L caused initial coughing and smarting of the eyes and nose, which went away after adaptation. 20 mg/L was just barely tolerable. Concentrations above 30 mg/L caused continuous coughing and tears, and concentrations above 40 mg/L were described as intolerable, suffocating, and impossible to bear for even short periods. Breathing air with concentration of 15 mg/L ethanol for 3 hours resulted in BACs from 0.2 to 4.5 g/L, depending on breathing rate.[83] It is not a particularly efficient or enjoyable method of becoming intoxicated.[4]
Ethanol is not absorbed significantly through intact skin. The steady state flux is 0.08 μmol/cm2/hr.[84] Applying a 70% ethanol solution to a skin area of 1000 cm2 for 1 hr would result in approximately 0.1 g of ethanol being absorbed.[85] The substantially increased levels of ethanol in the blood reported for some experiments are likely due to inadvertent inhalation.[4] A study that did not prevent respiratory uptake found that applying 200 mL of hand disinfectant containing 95% w/w ethanol (150 g ethanol total) over the course of 80 minutes in a 3-minutes-on 5-minutes-off pattern resulted in the median BAC among volunteers peaking 30 minutes after the last application at 17.5 mg/L (0.00175%). This BAC roughly corresponds to drinking one gram of pure ethanol.[86] Ethanol is rapidly absorbed through cut or damaged skin, with reports of ethanol intoxication and fatal poisoning.[87]
The timing of peak blood concentration varies depends on the type of alcoholic drink:[88]
- Vodka tonic: 36 ± 10 minutes after consumption
- Wine: 54 ± 14 minutes
- Beer: 62 ± 23 minutes
Also, carbonated alcoholic drinks seem to have a shorter onset compare to flat drinks in the same volume. One theory is that carbon dioxide in the bubbles somehow speeds the flow of alcohol into the intestines.[89]
Absorption is reduced by a large meal. Stress speeds up absorption.[81]
Distribution
[edit]After absorption, the alcohol goes through the portal vein to the liver, then through the hepatic veins to the heart, then the pulmonary arteries to the lungs, then the pulmonary veins to the heart again, and then enters systemic circulation.[13][90] Once in systematic circulation, ethanol distributes throughout the body, diffusing passively and crossing all biological membranes including the blood-brain barrier.[2][78] At equilibrium, ethanol is present in all body fluids and tissues in proportion to their water content. Ethanol does not bind to plasma proteins or other biomolecules.[13][2][3] The rate of distribution depends on blood supply,[4] specifically the cross-sectional area of the local capillary bed and the blood flow per gram of tissue.[13] As such, ethanol rapidly affects the brain, liver, and kidneys, which have high blood flow.[2] Other tissues with lower circulation, such as skeletal muscles and bone, require more time for ethanol to distribute into.[4][13] In rats, it takes around 10–15 minutes for tissue and venous blood to reach equilibrium.[91] Peak circulating levels of ethanol are usually reached within a range of 30 to 90 minutes of ingestion, with an average of 45 to 60 minutes.[4][2] People who have fasted overnight have been found to reach peak ethanol concentrations more rapidly, at within 30 minutes of ingestion.[4]
The volume of distribution Vd contributes about 15% of the uncertainty to Widmark's equation[92] and has been the subject of much research. Widmark originally used units of mass (g/kg) for EBAC, thus he calculated the apparent mass of distribution Md or mass of blood in kilograms. He fitted an equation of the body weight W in kg, finding an average rho-factor of 0.68 for men and 0.55 for women. This ρm has units of dose per body weight (g/kg) divided by concentration (g/kg) and is therefore dimensionless. However, modern calculations use weight/volume concentrations (g/L) for EBAC, so Widmark's rho-factors must be adjusted for the density of blood, 1.055 g/mL. This has units of dose per body weight (g/kg) divided by concentration (g/L blood) - calculation gives values of 0.64 L/kg for men and 0.52 L/kg for women, lower than the original.[93] Newer studies have updated these values to population-average ρv of 0.71 L/kg for men and 0.58 L/kg for women. But individual Vd values may vary significantly - the 95% range for ρv is 0.58-0.83 L/kg for males and 0.43-0.73 L/kg for females.[94] A more accurate method for calculating Vd is to use total body water (TBW) - experiments have confirmed that alcohol distributes almost exactly in proportion to TBW within the Widmark model.[95] TBW may be calculated using body composition analysis or estimated using anthropometric formulas based on age, height, and weight. Vd is then given by , where is the water content of blood, approximately 0.825 w/v for men and 0.838 w/v for women.[96]
These calculations assume Widmark's zero-order model for the effects of metabolization, and assume that TBW is almost exactly the volume of distribution of ethanol. Using a more complex model that accounts for non-linear metabolism, Norberg found that Vd was only 84-87% of TBW.[97] This finding was not reproduced in a newer study which found volumes of distribution similar to those in the literature.[79]
Metabolism
[edit]
Several metabolic pathways exist:
- One pathway involves alcohol dehydrogenase, particularly the IB (class I), beta polypeptide (ADH1B, EC 1.1.1.1) enzyme. The reaction uses NAD+ to convert the ethanol into acetaldehyde (a toxic carcinogen). The enzyme acetaldehyde dehydrogenase (aldehyde dehydrogenase 2 family ALDH2, EC 1.2.1.3) then converts the acetaldehyde into the non-toxic acetate ion (commonly found in acetic acid or vinegar).[4][6] This ion is in turn is broken down into carbon dioxide and water.[4] Specifically, acetate combines with coenzyme A (acetyl-CoA synthetase) to form acetyl-CoA, via the enzymes acyl-CoA synthetase short-chain family member 2 ACSS2 (EC 6.2.1.1) and acetyl-CoA synthase 2 (ACSS1). acetyl-CoA then participates in the citric acid cycle.[2][98] At even low physiological concentrations, ethanol completely saturates alcohol dehydrogenase.[4] This is because ethanol has high affinity for the enzyme and very high concentrations of ethanol occur when it is used as a recreational substance.[4]
- The microsomal ethanol-oxidizing system (MEOS), specifically mediated by the cytochrome P450 enzyme CYP2E1, is another major route of ethanol metabolism.[4][6] CYP2E1 is predominantly active at higher concentrations.[2] Repeated or chronic use of ethanol increases the activity of CYP2E1.[4][6]
- The activity of ADH and CYP2E1 alone does not appear sufficient to fully explain the increase in ethanol metabolism rate. There may be one or more additional pathways that metabolize as much as 25 to 35% of ethanol at typical concentrations.[3]
- A small amount of ethanol undergoes conjugation to form ethyl glucuronide and ethyl sulfate.[2]
Detailed ADH pathway
[edit]The reaction from ethanol to carbon dioxide and water proceeds in at least 11 steps in humans. C2H6O (ethanol) is converted to C2H4O (acetaldehyde), then to C2H4O2 (acetic acid), then to acetyl-CoA. Once acetyl-CoA is formed, it is free to enter directly into the citric acid cycle (TCA) and is converted to 2 CO2 molecules in 8 reactions. The equations:
- C2H6O(ethanol) + NAD+ → C2H4O(acetaldehyde) + NADH + H+
- C2H4O(acetaldehyde) + NAD+ + H2O → C2H4O2(acetic acid) + NADH + H+
- C2H4O2(acetic acid) + CoA + ATP → Acetyl-CoA + AMP + PPi
The Gibbs free energy is simply calculated from the free energy of formation of the product and reactants.[99][100] If catabolism of alcohol goes all the way to completion, then there is a very exothermic event yielding some 1325 kJ/mol of energy. If the reaction stops part way through the metabolic pathways, which happens because acetic acid is excreted in the urine after drinking, then not nearly as much energy can be derived from alcohol, indeed, only 215.1 kJ/mol. At the very least, the theoretical limits on energy yield are determined to be −215.1 kJ/mol to −1325.6 kJ/mol. The first with NADH is endothermic, requiring 47.2 kJ/mol of alcohol, or about 3 molecules of adenosine triphosphate (ATP) per molecule of ethanol.[original research?]
Variation
[edit]Variations in genes influence alcohol metabolism and drinking behavior.[101] Certain amino acid sequences in the enzymes used to oxidize ethanol are conserved (unchanged) going back to the last common ancestor over 3.5 bya.[102] Evidence suggests that humans evolved the ability to metabolize dietary ethanol between 7 and 21 million years ago, in a common ancestor shared with chimpanzees and gorillas but not orangutans.[103] Gene variation in these enzymes can lead to variation in catalytic efficiency between individuals. Some individuals have less effective metabolizing enzymes of ethanol, and can experience more marked symptoms from ethanol consumption than others.[104] However, those having acquired alcohol tolerance have a greater quantity of these enzymes, and metabolize ethanol more rapidly. Specifically, ethanol has been observed to be cleared more quickly by regular drinkers than non-drinkers.[104]
Falsely high BAC readings may be seen in patients with kidney or liver disease or failure. Such persons also have impaired acetaldehyde dehydrogenase, which causes acetaldehyde levels to peak higher, producing more severe hangovers and other effects such as flushing and tachycardia. Conversely, members of certain ethnicities that traditionally did not use alcoholic beverages have lower levels of alcohol dehydrogenases and thus "sober up" very slowly but reach lower aldehyde concentrations and have milder hangovers. The rate of detoxification of alcohol can also be slowed by certain drugs which interfere with the action of alcohol dehydrogenases, notably aspirin, furfural (which may be found in fusel alcohol), fumes of certain solvents, many heavy metals, and some pyrazole compounds. Also suspected of having this effect are cimetidine, ranitidine, and acetaminophen (paracetamol).[citation needed]
An "abnormal" liver with conditions such as hepatitis, cirrhosis, gall bladder disease, and cancer is likely to result in a slower rate of metabolism. People under 25 and women may process alcohol more slowly.[105]
Food such as fructose can increase the rate of alcohol metabolism. The effect can vary significantly from person to person, but a 100 g dose of fructose has been shown to increase alcohol metabolism by an average of 80%. In people with proteinuria and hematuria, fructose can cause falsely high BAC readings, due to kidney-liver metabolism.[106]
First-pass metabolism
[edit]During a typical drinking session, approximately 90% of the metabolism of ethanol occurs in the liver.[4][6] Alcohol dehydrogenase and aldehyde dehydrogenase are present at their highest concentrations (in liver mitochondria).[98][107] But these enzymes are widely expressed throughout the body, such as in the stomach and small intestine.[2] Some alcohol undergoes a first pass of metabolism in these areas, before it ever enters the bloodstream.[90]
In alcoholics
[edit]Under alcoholic conditions, the citric acid cycle is stalled by the oversupply of NADH derived from ethanol oxidation. The resulting backup of acetate shifts the reaction equilibrium for acetaldehyde dehydrogenase back towards acetaldehyde. Acetaldehyde subsequently accumulates and begins to form covalent bonds with cellular macromolecules, forming toxic adducts that, eventually, lead to death of the cell. This same excess of NADH from ethanol oxidation causes the liver to move away from fatty acid oxidation, which produces NADH, towards fatty acid synthesis, which consumes NADH. This consequent lipogenesis is believed to account largely for the pathogenesis of alcoholic fatty liver disease.
In human fetuses
[edit]In human embryos and fetuses, ethanol is not metabolized via ADH as ADH enzymes are not yet expressed to any significant quantity in human fetal liver (the induction of ADH only starts after birth, and requires years to reach adult levels).[108] Accordingly, the fetal liver cannot metabolize ethanol or other low molecular weight xenobiotics. In fetuses, ethanol is instead metabolized at much slower rates by different enzymes from the cytochrome P-450 superfamily (CYP), in particular by CYP2E1. The low fetal rate of ethanol clearance is responsible for the important observation that the fetal compartment retains high levels of ethanol long after ethanol has been cleared from the maternal circulation by the adult ADH activity in the maternal liver.[109] CYP2E1 expression and activity have been detected in various human fetal tissues after the onset of organogenesis (ca 50 days of gestation).[110] Exposure to ethanol is known to promote further induction of this enzyme in fetal and adult tissues. CYP2E1 is a major contributor to the so-called Microsomal Ethanol Oxidizing System (MEOS)[111] and its activity in fetal tissues is thought to contribute significantly to the toxicity of maternal ethanol consumption.[108][112] In presence of ethanol and oxygen, CYP2E1 is known[by whom?] to release superoxide radicals and induce the oxidation of polyunsaturated fatty acids to toxic aldehyde products like 4-hydroxynonenal (HNE).[citation needed]
The concentration of alcohol in breast milk produced during lactation is closely correlated to the individual's blood alcohol content.[113]
Elimination
[edit]Alcohol is removed from the bloodstream by a combination of metabolism, excretion, and evaporation. 90-98% of ingested ethanol is metabolized into carbon dioxide and water.[4] Around 5 to 10% of ethanol that is ingested is excreted unchanged in urine, breath, and sweat.[2] Transdermal alcohol that diffuses through the skin as insensible perspiration or is exuded as sweat (sensible perspiration) can be detected using wearable sensor technology[114] such as SCRAM ankle bracelet[115] or the more discreet ION Wearable.[116] Ethanol or its metabolites may be detectable in urine for up to 96 hours (3–5 days) after ingestion.[2]
Unlike most physiologically active materials, in typical recreational use, ethanol is removed from the bloodstream at an approximately constant rate (linear decay or zero-order kinetics), rather than at a rate proportional to the current concentration (exponential decay with a characteristic elimination half-life).[6][5] This is because typical doses of alcohol saturate the enzymes' capacity. In Widmark's model, the elimination rate from the blood, β, contributes 60% of the uncertainty.[92] Similarly to ρ, its value depends on the units used for blood.[93] β varies 58% by occasion and 42% between subjects; it is thus difficult to determine β precisely, and more practical to use a mean and a range of values. Typical elimination rates range from 10 to 34 mg/dL per hour,[6][4] with Jones recommending the range 0.10 - 0.25 g/L/h for forensic purposes, for all subjects.[117] Earlier studies found mean elimination rates of 15 mg/dL per hour for men and 18 mg/dL per hour for women,[6][4] but Jones found 0.148 g/L/h and 0.156 g/L/h respectively. Although the difference between sexes is statistically significant, it is small compared to the overall uncertainty, so Jones recommends using the value 0.15 for the mean for all subjects.[117] This mean rate is very roughly 8 grams of pure ethanol per hour (one British unit).[118] Explanations for the gender difference are quite varied and include liver size, secondary effects of the volume of distribution, and sex-specific hormones.[119] A 2023 study using a more complex two-compartment model with M-M elimination kinetics, with data from 60 men and 12 women, found statistically small effects of gender on maximal elimination rate and excluded them from the final model.[79]
At concentrations below 0.15-0.20 g/L, alcohol is eliminated more slowly and the elimination rate more closely follows first-order kinetics. The overall behavior of the elimination rate is described well by Michaelis–Menten kinetics. This change in behavior was not noticed by Widmark because he could not analyze low BAC levels.[93] The rate of elimination of ethanol is also increased at very high concentrations, such as in overdose, again more closely following first-order kinetics, with an elimination half-life of about 4 or 4.5 hours (a clearance rate of approximately 6 L/hour/70 kg). This is thought to be due to increased activity of CYP2E1.[3][2]
Eating food in proximity to drinking increases elimination rate significantly, mainly due to increased metabolism.[79]
Modeling
[edit]In fasting volunteers, blood levels of ethanol increase proportionally with the dose of ethanol administered.[78] Peak blood alcohol concentrations may be estimated by dividing the amount of ethanol ingested by the body weight of the individual and correcting for water dilution.[4] For time-dependent calculations, Swedish professor Erik Widmark developed a model of alcohol pharmacokinetics in the 1920s.[120] The model corresponds to a single-compartment model with instantaneous absorption and zero-order kinetics for elimination. The model is most accurate when used to estimate BAC a few hours after drinking a single dose of alcohol in a fasted state, and can be within 20% CV of the true value.[121][122] It is less accurate for BAC levels below 0.2 g/L (alcohol is not eliminated as quickly as predicted) and consumption with food (overestimating the peak BAC and time to return to zero).[123][93]
See also
[edit]References
[edit]- ^ Gilman JM, Ramchandani VA, Crouss T, Hommer DW (January 2012). "Subjective and neural responses to intravenous alcohol in young adults with light and heavy drinking patterns". Neuropsychopharmacology. 37 (2): 467–477. doi:10.1038/npp.2011.206. PMC 3242308. PMID 21956438.
- ^ a b c d e f g h i j k l m n o p q r s Principles of Addiction: Comprehensive Addictive Behaviors and Disorders. Academic Press. 17 May 2013. pp. 162–. ISBN 978-0-12-398361-9.
- ^ a b c d e f g Holford NH (November 1987). "Clinical pharmacokinetics of ethanol". Clinical Pharmacokinetics. 13 (5): 273–292. doi:10.2165/00003088-198713050-00001. PMID 3319346. S2CID 19723995.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y Pohorecky LA, Brick J (1988). "Pharmacology of ethanol". Pharmacology & Therapeutics. 36 (2–3): 335–427. doi:10.1016/0163-7258(88)90109-x. PMID 3279433.
- ^ a b Becker CE (September 1970). "The clinical pharmacology of alcohol". California Medicine. 113 (3): 37–45. PMC 1501558. PMID 5457514.
- ^ a b c d e f g h i Levine B (2003). Principles of Forensic Toxicology. Amer. Assoc. for Clinical Chemistry. pp. 161–. ISBN 978-1-890883-87-4.
- ^ Iber FL (26 November 1990). Alcohol and Drug Abuse as Encountered in Office Practice. CRC Press. pp. 74–. ISBN 978-0-8493-0166-7.
- ^ a b c Haynes WM, ed. (2011). CRC Handbook of Chemistry and Physics (92nd ed.). Boca Raton, Florida: CRC Press. p. 3.246. ISBN 1-4398-5511-0.
- ^ "Acetate, Ion chromatography standard solution, Safety Data Sheet". Thermo Fisher Scientific. 1 Apr 2024. p. 4.
- ^ Maxwell CR, Spangenberg RJ, Hoek JB, Silberstein SD, Oshinsky ML (December 2010). "Acetate causes alcohol hangover headache in rats". PLOS ONE. 5 (12): e15963. Bibcode:2010PLoSO...515963M. doi:10.1371/journal.pone.0015963. PMC 3013144. PMID 21209842.
- ^ 'Is coffee the real cure for a hangover?' by Bob Holmes, New Scientist, Jan. 15 2011, p. 17.
- ^ Jones AW (1991). "Forensic Science Aspects of Ethanol Metabolism". Forensic Science Progress 5. Vol. 5. pp. 31–89. doi:10.1007/978-3-642-58233-2_2. ISBN 978-3-540-53203-3.
- ^ a b c d e f g h i j Jones AW (September 2019). "Alcohol, its absorption, distribution, metabolism, and excretion in the body and pharmacokinetic calculations". WIREs Forensic Science. 1 (5). doi:10.1002/wfs2.1340.
- ^ Anstie FE (1874). "Final experiments on the elimination of alcohol from the body". The Practitioner. Vol. 13. John Brigg. pp. 15–28.
- ^ Atwater WO, Benedict FG (1902), "An Experimental Inquiry Regarding the Nutritive Value of Alcohol", Sixth Memoir, Memoirs of the National Academy of Sciences, vol. VIII, Washington: US Government Printing Office, pp. 231–397, S. Doc. 57-233
- ^ Widmark EM (1922). "Eine Mikromethode zur Bestimmung von Athylalkohol im Blut" [A micro-method for the determination of ethyl alcohol in the blood]. Biochemische Zeitschrift (in German). 131: 473–484. hdl:2027/uc1.b3778068.
- ^ "Die theoretischen Grundlagen und die praktische Verwendbarkeit der gerichtlich-medizinischen Alkoholbestimmung" [Principles and applications of medicolegal alcohol determination]. Journal of the American Medical Association (book review). 98 (21): 1834. 21 May 1932. doi:10.1001/jama.1932.02730470056035.
- ^ Watson PE, Watson ID, Batt RD (July 1981). "Prediction of blood alcohol concentrations in human subjects. Updating the Widmark Equation". Journal of Studies on Alcohol. 42 (7): 547–556. doi:10.15288/jsa.1981.42.547. PMID 7289599.
- ^ Maskell PD, Jones AW, Heymsfield SB, Shapses S, Johnston A (November 2020). "Total body water is the preferred method to use in forensic blood-alcohol calculations rather than ethanol's volume of distribution". Forensic Science International. 316: 110532. doi:10.1016/j.forsciint.2020.110532. PMID 33099270.
- ^ a b c d e f g h i j k Lobo IA, Harris RA (July 2008). "GABA(A) receptors and alcohol". Pharmacology, Biochemistry, and Behavior. 90 (1): 90–94. doi:10.1016/j.pbb.2008.03.006. PMC 2574824. PMID 18423561.
- ^ a b c d e f g h i j k l m n o p q r s t u Santhakumar V, Wallner M, Otis TS (May 2007). "Ethanol acts directly on extrasynaptic subtypes of GABAA receptors to increase tonic inhibition". Alcohol. 41 (3): 211–221. doi:10.1016/j.alcohol.2007.04.011. PMC 2040048. PMID 17591544.
- ^ a b c d e Spanagel R (April 2009). "Alcoholism: a systems approach from molecular physiology to addictive behavior". Physiological Reviews. 89 (2): 649–705. doi:10.1152/physrev.00013.2008. PMID 19342616.
- ^ Lovinger DM, Roberto M (2013). "Synaptic effects induced by alcohol". Current Topics in Behavioral Neurosciences. 13: 31–86. doi:10.1007/7854_2011_143. ISBN 978-3-642-28719-0. PMC 4791588. PMID 21786203.
- ^ Lovinger DM, Homanics GE (May 2007). "Tonic for what ails us? high-affinity GABAA receptors and alcohol". Alcohol. 41 (3): 139–143. doi:10.1016/j.alcohol.2007.03.008. PMC 2043151. PMID 17521844.
- ^ a b Vengeliene V, Bilbao A, Molander A, Spanagel R (May 2008). "Neuropharmacology of alcohol addiction". British Journal of Pharmacology. 154 (2): 299–315. doi:10.1038/bjp.2008.30. PMC 2442440. PMID 18311194.
- ^ a b c d Narahashi T, Kuriyama K, Illes P, Wirkner K, Fischer W, Mühlberg K, et al. (May 2001). "Neuroreceptors and ion channels as targets of alcohol". Alcoholism: Clinical and Experimental Research. 25 (5 Suppl ISBRA): 182S–188S. doi:10.1097/00000374-200105051-00030. PMID 11391069.
- ^ a b c Olsen RW, Li GD, Wallner M, Trudell JR, Bertaccini EJ, Lindahl E, et al. (March 2014). "Structural models of ligand-gated ion channels: sites of action for anesthetics and ethanol". Alcoholism: Clinical and Experimental Research. 38 (3): 595–603. doi:10.1111/acer.12283. PMC 3959612. PMID 24164436.
- ^ Chung HW, Petersen EN, Cabanos C, Murphy KR, Pavel MA, Hansen AS, et al. (January 2019). "A Molecular Target for an Alcohol Chain-Length Cutoff". Journal of Molecular Biology. 431 (2): 196–209. doi:10.1016/j.jmb.2018.11.028. PMC 6360937. PMID 30529033.
- ^ Robinson CV, Rohacs T, Hansen SB (September 2019). "Tools for Understanding Nanoscale Lipid Regulation of Ion Channels". Trends in Biochemical Sciences. 44 (9): 795–806. doi:10.1016/j.tibs.2019.04.001. PMC 6729126. PMID 31060927.
- ^ Laverty D, Desai R, Uchański T, Masiulis S, Stec WJ, Malinauskas T, et al. (January 2019). "Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer". Nature. 565 (7740): 516–520. Bibcode:2019Natur.565..516L. doi:10.1038/s41586-018-0833-4. PMC 6364807. PMID 30602789.
- ^ a b c d e Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 15: Reinforcement and Addictive Disorders". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. p. 372. ISBN 978-0-07-148127-4.
- ^ Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, et al. (September 1997). "Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors". Nature. 389 (6649): 385–389. Bibcode:1997Natur.389..385M. doi:10.1038/38738. PMID 9311780. S2CID 4393717.
- ^ a b c d Möykkynen T, Korpi ER (July 2012). "Acute effects of ethanol on glutamate receptors". Basic & Clinical Pharmacology & Toxicology. 111 (1): 4–13. doi:10.1111/j.1742-7843.2012.00879.x. PMID 22429661.
- ^ a b Lovinger DM (August 1999). "5-HT3 receptors and the neural actions of alcohols: an increasingly exciting topic". Neurochemistry International. 35 (2): 125–130. doi:10.1016/S0197-0186(99)00054-6. PMID 10405996. S2CID 1391767.
- ^ a b Söderpalm B, Lidö HH, Ericson M (November 2017). "The Glycine Receptor-A Functionally Important Primary Brain Target of Ethanol". Alcoholism: Clinical and Experimental Research. 41 (11): 1816–1830. doi:10.1111/acer.13483. PMID 28833225.
- ^ Narahashi T, Aistrup GL, Marszalec W, Nagata K (August 1999). "Neuronal nicotinic acetylcholine receptors: a new target site of ethanol". Neurochemistry International. 35 (2): 131–141. doi:10.1016/S0197-0186(99)00055-8. PMID 10405997. S2CID 40991187.
- ^ Wu J, Gao M, Taylor DH (March 2014). "Neuronal nicotinic acetylcholine receptors are important targets for alcohol reward and dependence". Acta Pharmacologica Sinica. 35 (3): 311–315. doi:10.1038/aps.2013.181. PMC 4647894. PMID 24464050.
- ^ Steffensen SC, Shin SI, Nelson AC, Pistorius SS, Williams SB, Woodward TJ, et al. (September 2018). "α6 subunit-containing nicotinic receptors mediate low-dose ethanol effects on ventral tegmental area neurons and ethanol reward". Addiction Biology. 23 (5): 1079–1093. doi:10.1111/adb.12559. PMC 5849490. PMID 28901722.
- ^ Sitte H, Freissmuth M (2 August 2006). Neurotransmitter Transporters. Springer Science & Business Media. pp. 472–. ISBN 978-3-540-29784-0.
- ^ Allen-Gipson DS, Jarrell JC, Bailey KL, Robinson JE, Kharbanda KK, Sisson JH, et al. (May 2009). "Ethanol blocks adenosine uptake via inhibiting the nucleoside transport system in bronchial epithelial cells". Alcoholism: Clinical and Experimental Research. 33 (5): 791–798. doi:10.1111/j.1530-0277.2009.00897.x. PMC 2940831. PMID 19298329.
- ^ a b Wang X, Wang G, Lemos JR, Treistman SN (September 1994). "Ethanol directly modulates gating of a dihydropyridine-sensitive Ca2+ channel in neurohypophysial terminals". The Journal of Neuroscience. 14 (9): 5453–5460. doi:10.1523/JNEUROSCI.14-09-05453.1994. PMC 6577079. PMID 7521910.
- ^ Dopico AM, Bukiya AN, Kuntamallappanavar G, Liu J (2016). "Modulation of BK Channels by Ethanol". International Review of Neurobiology. 128: 239–279. doi:10.1016/bs.irn.2016.03.019. ISBN 978-0-12-803619-8. PMC 5257281. PMID 27238266.
- ^ Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T, et al. (December 1999). "Ethanol opens G-protein-activated inwardly rectifying K+ channels". Nature Neuroscience. 2 (12): 1091–1097. doi:10.1038/16019. PMID 10570486. S2CID 28730710.
- ^ Situmorang JH, Lin HH, Lo H, Lai CC (January 2018). "Role of neuronal nitric oxide synthase (nNOS) at medulla in tachycardia induced by repeated administration of ethanol in conscious rats". Journal of Biomedical Science. 25 (1): 8. doi:10.1186/s12929-018-0409-5. PMC 5791364. PMID 29382335.
- ^ a b Melis M, Enrico P, Peana AT, Diana M (November 2007). "Acetaldehyde mediates alcohol activation of the mesolimbic dopamine system". The European Journal of Neuroscience. 26 (10): 2824–2833. doi:10.1111/j.1460-9568.2007.05887.x. PMID 18001279. S2CID 25110014.
- ^ a b c Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS, et al. (September 2009). "The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress". Psychopharmacology. 205 (4): 529–564. doi:10.1007/s00213-009-1562-z. PMC 2814770. PMID 19455309.
- ^ a b c d Wallner M, Olsen RW (May 2008). "Physiology and pharmacology of alcohol: the imidazobenzodiazepine alcohol antagonist site on subtypes of GABAA receptors as an opportunity for drug development?". British Journal of Pharmacology. 154 (2): 288–298. doi:10.1038/bjp.2008.32. PMC 2442438. PMID 18278063.
- ^ a b c d e f g h Harrison NL, Skelly MJ, Grosserode EK, Lowes DC, Zeric T, Phister S, et al. (August 2017). "Effects of acute alcohol on excitability in the CNS". Neuropharmacology. 122: 36–45. doi:10.1016/j.neuropharm.2017.04.007. PMC 5657304. PMID 28479395.
- ^ Wallner M, Hanchar HJ, Olsen RW (December 2003). "Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans". Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15218–15223. Bibcode:2003PNAS..10015218W. doi:10.1073/pnas.2435171100. PMC 299963. PMID 14625373.
- ^ a b Baur R, Kaur KH, Sigel E (December 2009). "Structure of alpha6 beta3 delta GABA(A) receptors and their lack of ethanol sensitivity". Journal of Neurochemistry. 111 (5): 1172–1181. doi:10.1111/j.1471-4159.2009.06387.x. PMID 19765192. S2CID 10577529.
- ^ Förstera B, Castro PA, Moraga-Cid G, Aguayo LG (2016). "Potentiation of Gamma Aminobutyric Acid Receptors (GABAAR) by Ethanol: How Are Inhibitory Receptors Affected?". Frontiers in Cellular Neuroscience. 10: 114. doi:10.3389/fncel.2016.00114. PMC 4858537. PMID 27199667.
- ^ Tobin V, Leng G, Ludwig M (2012). "The involvement of actin, calcium channels and exocytosis proteins in somato-dendritic oxytocin and vasopressin release". Frontiers in Physiology. 3: 261. doi:10.3389/fphys.2012.00261. PMC 3429037. PMID 22934017.
- ^ Chiodera P, Coiro V (May 1990). "Inhibitory effect of ethanol on the arginine vasopressin response to insulin-induced hypoglycemia and the role of endogenous opioids". Neuroendocrinology. 51 (5): 501–504. doi:10.1159/000125383. PMID 2112727.
- ^ Treistman SN, Bayley H, Lemos JR, Wang XM, Nordmann JJ, Grant AJ (1991). "Effects of ethanol on calcium channels, potassium channels, and vasopressin release". Annals of the New York Academy of Sciences. 625 (1): 249–263. Bibcode:1991NYASA.625..249T. doi:10.1111/j.1749-6632.1991.tb33844.x. PMID 1647726. S2CID 28281696.
- ^ Walter HJ, Messing RO (August 1999). "Regulation of neuronal voltage-gated calcium channels by ethanol". Neurochemistry International. 35 (2): 95–101. doi:10.1016/s0197-0186(99)00050-9. PMID 10405992. S2CID 36172178.
- ^ Katsura M, Shibasaki M, Hayashida S, Torigoe F, Tsujimura A, Ohkuma S (October 2006). "Increase in expression of alpha1 and alpha2/delta1 subunits of L-type high voltage-gated calcium channels after sustained ethanol exposure in cerebral cortical neurons". Journal of Pharmacological Sciences. 102 (2): 221–230. doi:10.1254/jphs.fp0060781. PMID 17031067.
- ^ Malysz J, Afeli SA, Provence A, Petkov GV (January 2014). "Ethanol-mediated relaxation of guinea pig urinary bladder smooth muscle: involvement of BK and L-type Ca2+ channels". American Journal of Physiology. Cell Physiology. 306 (1): C45–C58. doi:10.1152/ajpcell.00047.2013. PMC 3919972. PMID 24153429.
- ^ Cueva JP, Giorgioni G, Grubbs RA, Chemel BR, Watts VJ, Nichols DE (November 2006). "trans-2,3-dihydroxy-6a,7,8,12b-tetrahydro-6H-chromeno[3,4-c]isoquinoline: synthesis, resolution, and preliminary pharmacological characterization of a new dopamine D1 receptor full agonist". Journal of Medicinal Chemistry. 49 (23): 6848–6857. doi:10.1021/jm0604979. PMID 17154515.
- ^ Michaelides MR, Hong Y, DiDomenico S, Asin KE, Britton DR, Lin CW, et al. (September 1995). "(5aR,11bS)-4,5,5a,6,7,11b-hexahydro-2-propyl-3-thia-5-azacyclopent-1- ena[c]-phenanthrene-9,10-diol (A-86929): a potent and selective dopamine D1 agonist that maintains behavioral efficacy following repeated administration and characterization of its diacetyl prodrug (ABT-431)". Journal of Medicinal Chemistry. 38 (18): 3445–3447. doi:10.1021/jm00018a002. PMID 7658429.
- ^ Karahanian E, Quintanilla ME, Tampier L, Rivera-Meza M, Bustamante D, Gonzalez-Lira V, et al. (April 2011). "Ethanol as a prodrug: brain metabolism of ethanol mediates its reinforcing effects". Alcoholism: Clinical and Experimental Research. 35 (4): 606–612. doi:10.1111/j.1530-0277.2011.01439.x. PMC 3142559. PMID 21332529.
- ^ a b c d e "Alcoholism – Homo sapiens (human) Database entry". KEGG Pathway. 29 October 2014. Retrieved 9 February 2015.
- ^ a b c d e f g h Kanehisa Laboratories (29 October 2014). "Alcoholism – Homo sapiens (human)". KEGG Pathway. Retrieved 31 October 2014.
- ^ Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". The American Journal of Drug and Alcohol Abuse. 40 (6): 428–437. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
- ^ Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues in Clinical Neuroscience. 15 (4): 431–443. doi:10.31887/DCNS.2013.15.4/enestler. PMC 3898681. PMID 24459410.
Despite the Importance of Numerous Psychosocial Factors, at its Core, Drug Addiction Involves a Biological Process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. ... A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal's sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement
- ^ Robison AJ, Nestler EJ (October 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews. Neuroscience. 12 (11): 623–637. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
- ^ Pohorecky LA, Brick J (1988). "Pharmacology of ethanol". Pharmacology & Therapeutics. 36 (2–3): 335–427. doi:10.1016/0163-7258(88)90109-X. PMID 3279433.
- ^ a b Liu Y, Hunt WA (6 December 2012). The "Drunken" Synapse: Studies of Alcohol-Related Disorders. Springer Science & Business Media. pp. 40–. ISBN 978-1-4615-4739-6.
- ^ Rubin R, Strayer DS, Rubin E, McDonald JM (2008). Rubin's Pathology: Clinicopathologic Foundations of Medicine. Lippincott Williams & Wilkins. pp. 257–. ISBN 978-0-7817-9516-6.
- ^ The World Health Organization (2007) Alcohol and Injury in Emergency Departments
- ^ "Fatty Acid Synthesis".
- ^ "Glycerolipid Metabolism".
- ^ "Bile Acid Biosynthesis".
- ^ Fath BD, Jørgensen SE (23 August 2018). Encyclopedia of dcology (Second ed.). Amsterdam, Netherlands: Elsevier. ISBN 978-0-444-64130-4. OCLC 1054599976.
- ^ Mendez ML (2016). Electronic noses and tongues in food science. London: Academic Press. ISBN 978-0-12-800402-9. OCLC 940961460.
- ^ Tillonen J. Ethanol, acetaldehyde and gastrointestinal flora (PDF) (Ph.D. thesis). Helsingin Yliopisto.
- ^ Ostrovsky Y (July 1986). "Endogenous ethanol--its metabolic, behavioral and biomedical significance". Alcohol. 3 (4): 239–247. doi:10.1016/0741-8329(86)90032-7. PMID 3530279.
- ^ Logan BK, Jones AW (July 2000). "Endogenous ethanol 'auto-brewery syndrome' as a drunk-driving defence challenge". Medicine, Science, and the Law. 40 (3): 206–215. doi:10.1177/002580240004000304. PMID 10976182. S2CID 6926029.
- ^ a b c d e Henri B, Kissin B (1996). The Pharmacology of Alcohol and Alcohol Dependence. Oxford University Press. pp. 18–. ISBN 978-0-19-510094-5.
- ^ a b c d Büsker S, Jones AW, Hahn RG, Taubert M, Klotz U, Schwab M, et al. (June 2023). "Population Pharmacokinetics as a Tool to Reevaluate the Complex Disposition of Ethanol in the Fed and Fasted States". Journal of Clinical Pharmacology. 63 (6): 681–694. doi:10.1002/jcph.2205. PMID 36688276.
- ^ Berggren SM, Goldberg L (March 1940). "The Absorption of Ethyl Alcohol from the Gastro-Intestinal Tract as a Diffusion Process". Acta Physiologica Scandinavica. 1 (3): 246–270. doi:10.1111/j.1748-1716.1940.tb00272.x.
- ^ a b "Absorption Rate Factors". BHS.UMN.edu. Archived from the original on 18 January 2013. Retrieved 6 March 2018.
When food is ingested, the pyloric valve at the bottom of the stomach will close in order to hold food in the stomach for digestion and thus keep the alcohol from reaching the small intestine. The larger the meal and closer in time to drinking, the lower the peak of alcohol concentration; some studies indicate up to a 20% reduction in peak blood alcohol level.
Stress causes the stomach to empty directly into the small intestine, where alcohol is absorbed even faster.
Liquor mixed with soda or other bubbly drinks speeds up the passage of alcohol from the stomach to the small intestine, which increases the speed of absorption. - ^ Gréhant N (1897). "Absorption par les poumons de vapeur d'alcool mélangée avec l'air" [Absorption by the lungs of alcohol vapor mixed with air]. Bulletin du Muséum d'histoire naturelle (in French). 3 (1): 28–29.
- ^ Lester D, Greenberg LA (June 1951). "The inhalation of ethyl alcohol by man. I. Industrial hygiene and medicolegal aspects. II. Individuals treated with tetraethylthiuram disulfide" (PDF). Quarterly Journal of Studies on Alcohol. 12 (2): 168–178. doi:10.15288/qjsa.1951.12.167. PMID 14844643.
- ^ Scheuplein RJ, Blank IH (May 1973). "Mechanism of percutaneous absorption. IV. Penetration of nonelectrolytes (alcohols) from aqueous solutions and from pure liquids". The Journal of Investigative Dermatology. 60 (5): 286–296. doi:10.1111/1523-1747.ep12723090. PMID 4758734.
- ^ Schaefer H, Redelmeier TE (26 September 1996). Skin Barrier: Principles of Percutaneous Absorption. p. 247. doi:10.1159/000425546.
- ^ Kramer A, Below H, Bieber N, Kampf G, Toma CD, Huebner NO, et al. (October 2007). "Quantity of ethanol absorption after excessive hand disinfection using three commercially available hand rubs is minimal and below toxic levels for humans". BMC Infectious Diseases. 7 (1): 117. doi:10.1186/1471-2334-7-117. PMC 2089071. PMID 17927841.
- ^ Lachenmeier DW (November 2008). "Safety evaluation of topical applications of ethanol on the skin and inside the oral cavity". Journal of Occupational Medicine and Toxicology. 3: 26. doi:10.1186/1745-6673-3-26. PMC 2596158. PMID 19014531.
- ^ Mitchell MC, Teigen EL, Ramchandani VA (May 2014). "Absorption and peak blood alcohol concentration after drinking beer, wine, or spirits". Alcoholism: Clinical and Experimental Research. 38 (5): 1200–1204. doi:10.1111/acer.12355. PMC 4112772. PMID 24655007.
- ^ "Champagne does get you drunk faster". New Scientist.
- ^ a b Alan J.Buglass, ed. (2011). Handbook of alcoholic beverages : technical, analytical and nutritional aspects. Chichester: Wiley. ISBN 978-0-470-97665-4. Retrieved 6 July 2013.
- ^ Sunahara GI, Kalant H, Schofield M, Grupp L (December 1978). "Regional distribution of ethanol in the rat brain". Canadian Journal of Physiology and Pharmacology. 56 (6): 988–992. doi:10.1139/y78-157. PMID 743637.
- ^ a b Maskell PD, Cooper GA (September 2020). "The Contribution of Body Mass and Volume of Distribution to the Estimated Uncertainty Associated with the Widmark Equation". Journal of Forensic Sciences. 65 (5): 1676–1684. doi:10.1111/1556-4029.14447. PMID 32421216. S2CID 218677989.
- ^ a b c d Jones AW (July 2011). "Pharmacokinetics of Ethanol - Issues of Forensic Importance". Forensic Science Review. 23 (2): 91–136. PMID 26231237.
- ^ Maskell PD, Heymsfield SB, Shapses S, Limoges JF (September 2023). "Population ranges for the volume of distribution (Vd ) of alcohol for use in forensic alcohol calculations". Journal of Forensic Sciences. 68 (5): 1843–1845. doi:10.1111/1556-4029.15317. PMID 37345356.
- ^ Endres HG, Grüner O (November 1994). "Comparison of D2O and ethanol dilutions in total body water measurements in humans". The Clinical Investigator. 72 (11): 830–837. doi:10.1007/BF00190736. PMID 7894207.
- ^ Maskell PD, Jones AW, Heymsfield SB, Shapses S, Johnston A (November 2020). "Total body water is the preferred method to use in forensic blood-alcohol calculations rather than ethanol's volume of distribution". Forensic Science International. 316: 110532. doi:10.1016/j.forsciint.2020.110532. PMID 33099270. S2CID 224966411.
- ^ Norberg A, Sandhagen B, Bratteby LE, Gabrielsson J, Jones AW, Fan H, et al. (October 2001). "Do ethanol and deuterium oxide distribute into the same water space in healthy volunteers?". Alcoholism: Clinical and Experimental Research. 25 (10): 1423–1430. doi:10.1111/j.1530-0277.2001.tb02143.x. PMID 11696661.
- ^ a b Smith, C., Marks, Allan D., Lieberman, Michael, 2005, Marks' Basic Medical Biochemistry: A Clinical Approach, 2nd ed., Lippincott Williams & Wilkins, USA, p. 458
- ^ CRC Handbook of Chemistry and Physics, 81st Edition, 2000
- ^ "MetaCyc EC 6.2.1.1".
- ^ Agarwal DP (November 2001). "Genetic polymorphisms of alcohol metabolizing enzymes". Pathologie-Biologie. 49 (9): 703–709. doi:10.1016/s0369-8114(01)00242-5. PMID 11762132.
- ^ NIH/NLM/NCBI/IEB/CDD. "NCBI CDD Conserved Protein Domain ADH_zinc_N". www.ncbi.nlm.nih.gov. Retrieved 2018-04-28.
- ^ Carrigan MA, Uryasev O, Frye CB, Eckman BL, Myers CR, Hurley TD, et al. (January 2015). "Hominids adapted to metabolize ethanol long before human-directed fermentation". Proceedings of the National Academy of Sciences of the United States of America. 112 (2): 458–463. Bibcode:2015PNAS..112..458C. doi:10.1073/pnas.1404167111. PMC 4299227. PMID 25453080.
- ^ a b Agarwal DP, Goedde HW (April 1992). "Pharmacogenetics of alcohol metabolism and alcoholism". Pharmacogenetics. 2 (2): 48–62. doi:10.1097/00008571-199204000-00002. PMID 1302043.
- ^ Thomasson HR (2002). "Gender Differences in Alcohol Metabolism". Recent Developments in Alcoholism. Vol. 12. Plenum Press. pp. 163–72. doi:10.1007/0-306-47138-8_9. ISBN 978-0-306-44921-5. PMID 7624539.
- ^ Fructose & ethanol[improper synthesis?]
- Carpenter TM, Lee RC (1937). "The effect of fructose on the metabolism of ethyl alcohol in man". Journal of Pharmacology and Experimental Therapeutics. 60 (3). Retrieved 23 June 2016.
- Tygstrup N, Winkler K, Lundquist F (May 1965). "The Mechanism of the Fructose Effect on the Ethanol Metabolism of the Human Liver". The Journal of Clinical Investigation. 44 (5): 817–830. doi:10.1172/JCI105194. PMC 292558. PMID 14276139.
- Patel AR, Paton AM, Rowan T, Lawson DH, Linton AL (August 1969). "Clinical studies on the effect of laevulose on the rate of metabolism of ethyl alcohol". Scottish Medical Journal. 14 (8): 268–271. doi:10.1177/003693306901400803. PMID 5812044. S2CID 3067691.
- Lowenstein LM, Simone R, Boulter P, Nathan P (September 1970). "Effect of fructose on alcohol concentrations in the blood in man". JAMA. 213 (11): 1899–1901. doi:10.1001/jama.1970.03170370083021. PMID 4318655.
- Pawan GL (September 1972). "Metabolism of alcohol (ethanol) in man". The Proceedings of the Nutrition Society. 31 (2): 83–89. doi:10.1079/pns19720020. PMID 4563296.
- Thieden HI, Grunnet N, Damgaard SE, Sestoft L (October 1972). "Effect of fructose and glyceraldehyde on ethanol metabolism in human liver and in rat liver". European Journal of Biochemistry. 30 (2): 250–261. doi:10.1111/j.1432-1033.1972.tb02093.x. PMID 4145889.
- Soterakis J, Iber FL (March 1975). "Increased rate of alcohol removal from blood with oral fructose and sucrose". The American Journal of Clinical Nutrition. 28 (3): 254–257. doi:10.1093/ajcn/28.3.254. PMID 1119423.
- Rawat AK (February 1977). "Effects of fructose and other substances on ethanol and acetaldehyde metabolism in man". Research Communications in Chemical Pathology and Pharmacology. 16 (2): 281–290. PMID 847286.
- Iber FL (September 1977). "The effect of fructose on alcohol metabolism". Archives of Internal Medicine. 137 (9): 1121. doi:10.1001/archinte.137.9.1121. PMID 901079.
- Bode JC, Bode C, Thiele D (February 1979). "Alcohol metabolism in man: effect of intravenous fructose infusion on blood ethanol elimination rate following stimulation by phenobarbital treatment or chronic alcohol consumption". Klinische Wochenschrift. 57 (3): 125–130. doi:10.1007/bf01476052. PMID 439778. S2CID 8813046.
- Sprandel U, Tröger HD, Liebhardt EW, Zöllner N (1980). "Acceleration of ethanol elimination with fructose in man". Nutrition and Metabolism. 24 (5): 324–330. doi:10.1159/000176278. PMID 7443107.
- Meyer BH, Müller FO, Hundt HK (November 1982). "The effect of fructose on blood alcohol levels in man". South African Medical Journal = Suid-Afrikaanse Tydskrif vir Geneeskunde. 62 (20): 719–721. PMID 6753183.
- Crownover BP, La Dine J, Bradford B, Glassman E, Forman D, Schneider H, et al. (March 1986). "Activation of ethanol metabolism in humans by fructose: importance of experimental design". The Journal of Pharmacology and Experimental Therapeutics. 236 (3): 574–579. PMID 3950864.
- Mascord D, Smith J, Starmer GA, Whitfield JB (1991). "The effect of fructose on alcohol metabolism and on the [lactate]/[pyruvate] ratio in man". Alcohol and Alcoholism. 26 (1): 53–59. PMID 1854373.
- Onyesom I, Anosike EO (June 2004). "Oral fructose-induced changes in blood ethanol oxidokinetic data among healthy Nigerians". The Southeast Asian Journal of Tropical Medicine and Public Health. 35 (2): 476–480. PMID 15691159.
- Uzuegbu UE, Onyesom I (June 2009). "Fructose-induced increase in ethanol metabolism and the risk of Syndrome X in man". Comptes Rendus Biologies. 332 (6): 534–538. doi:10.1016/j.crvi.2009.01.007. PMID 19520316.
- ^ Tanaka F, Shiratori Y, Yokosuka O, Imazeki F, Tsukada Y, Omata M (June 1997). "Polymorphism of alcohol-metabolizing genes affects drinking behavior and alcoholic liver disease in Japanese men". Alcoholism: Clinical and Experimental Research. 21 (4): 596–601. doi:10.1111/j.1530-0277.1997.tb03808.x. PMID 9194910.
- ^ a b Ernst van Faassen and Onni Niemelä, Biochemistry of prenatal alcohol exposure, NOVA Science Publishers, New York 2011.[page needed]
- ^ Nava-Ocampo AA, Velázquez-Armenta Y, Brien JF, Koren G (June 2004). "Elimination kinetics of ethanol in pregnant women". Reproductive Toxicology. 18 (4): 613–617. Bibcode:2004RepTx..18..613N. doi:10.1016/j.reprotox.2004.02.012. PMID 15135856.
- ^ Brzezinski MR, Boutelet-Bochan H, Person RE, Fantel AG, Juchau MR (June 1999). "Catalytic activity and quantitation of cytochrome P-450 2E1 in prenatal human brain". The Journal of Pharmacology and Experimental Therapeutics. 289 (3): 1648–1653. PMID 10336564.
- ^ Lieber CS (October 2004). "The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role". Drug Metabolism Reviews. 36 (3–4): 511–529. doi:10.1081/dmr-200033441. PMID 15554233. S2CID 27992318.
- ^ Pregnancy and Alcohol Consumption, ed. J.D. Hoffmann, NOVA Science Publishers, New York 2011.[page needed]
- ^ Haastrup MB, Pottegård A, Damkier P (February 2014). "Alcohol and breastfeeding". Basic & Clinical Pharmacology & Toxicology. 114 (2): 168–173. doi:10.1111/bcpt.12149. PMID 24118767.
- ^ Lansdorp B, Ramsay W, Hamidand R, Strenk E (May 2019). "Wearable Enzymatic Alcohol Biosensor". Sensors. 19 (10): 2380. Bibcode:2019Senso..19.2380L. doi:10.3390/s19102380. PMC 6566815. PMID 31137611.
- ^ "SCRAM CAM® Bracelet Alcohol Ankle Monitor". SCRAM Systems. Retrieved 2022-03-19.
- ^ "ION Wearable". ION Wearable. Retrieved 2022-03-19.
- ^ a b Jones AW (July 2010). "Evidence-based survey of the elimination rates of ethanol from blood with applications in forensic casework". Forensic Science International. 200 (1–3): 1–20. doi:10.1016/j.forsciint.2010.02.021. PMID 20304569.
- ^ "How long does alcohol stay in your blood?". NHS Choices. 26 June 2018.
- ^ Dettling A, Skopp G, Graw M, Haffner HT (May 2008). "The influence of sex hormones on the elimination kinetics of ethanol". Forensic Science International. 177 (2–3): 85–89. doi:10.1016/j.forsciint.2007.11.002. PMID 18079079.
- ^ Kuwatch E. "Fast Eddie's 8/10 Method of Hand Calculating Blood Alcohol Concentration: A Simple Method For Using Widmark's Formula". Archived from the original on 2003-12-02.
- ^ Zuba D, Piekoszewski W (2004). "Uncertainty in Theoretical Calculations of Alcohol Concentration". Proc. 17th Internat. Conf. on Alcohol, Drugs and Traffic Safety.
- ^ Gullberg RG (October 2007). "Estimating the uncertainty associated with Widmark's equation as commonly applied in forensic toxicology". Forensic Science International. 172 (1): 33–39. doi:10.1016/j.forsciint.2006.11.010. PMID 17210238.
- ^ Searle J (January 2015). "Alcohol calculations and their uncertainty". Medicine, Science, and the Law. 55 (1): 58–64. doi:10.1177/0025802414524385. PMC 4361698. PMID 24644224.
- 5-HT3 agonists
- AMPA receptor antagonists
- Adenosine reuptake inhibitors
- Alcohol
- Alcohol abuse
- Alcohol and health
- Alcohol dehydrogenase inhibitors
- Alcohol law
- Alcohols
- Analgesics
- Anaphrodisia
- Anxiolytics
- Calcium channel blockers
- Depressogens
- Diuretics
- Drugs acting on the nervous system
- Drugs with unknown mechanisms of action
- Emetics
- Ethanol
- Euphoriants
- GABAA receptor positive allosteric modulators
- General anesthetics
- Glycine reuptake inhibitors
- Hepatotoxins
- Human metabolites
- Hypnotics
- IARC Group 1 carcinogens
- Kainate receptor antagonists
- NMDA receptor antagonists
- Neurotoxins
- Nicotinic agonists
- Ototoxicity
- Psychoactive drugs
- Sedatives
- Teratogens