Malaltia de Charcot-Marie-Tooth
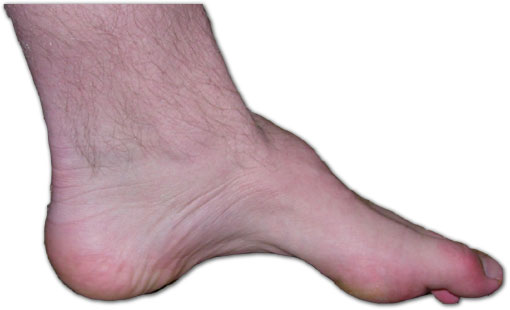 Peu d'una persona amb la malaltia de Charcot-Marie-Tooth. La manca de múscul, la planta del peu arquejada i els dits de peu en martell són signes d'aquesta malaltia genètica. | |
| Tipus | neuropatia motora perifèrica hereditària, malaltia neuromuscular, malaltia monogènica i malaltia |
|---|---|
| Epònim | Jean-Martin Charcot, Pierre Marie i Howard Henry Tooth (en) |
| Especialitat | neurologia |
| Clínica-tractament | |
| Símptomes | debilitat muscular, atròfia muscular, peu buit, pèrdua sensorial, hyporeflexia (en) |
| Exàmens | prova genètica, tècniques de diagnòstic neurològic i electromiografia |
| Tractament | desconegut i fisioteràpia |
| Patogènesi | |
| Associació genètica | PMP22 (en) |
| Causat per | mutació |
| Classificació | |
| CIM-10 | G60.0 |
| CIM-9 | 356.1 |
| Recursos externs | |
| OMIM | 311860 |
| DiseasesDB | 5815 2343 |
| MedlinePlus | 000727 |
| eMedicine | orthoped/43 pmr/29 |
| MeSH | D002607 |
| Orphanet | 166 |
| UMLS CUI | C0007959 |
| DOID | DOID:10595 |
La malaltia de Charcot-Marie-Tooth (CMT) és un dels trastorns desmielinitzants hereditaris més comuns que afecta aproximadament a 1 de cada 2.500 persones als Estats Units. La malaltia rep el nom dels tres metges que la van identificar per primera vegada en 1886 -Jean-Marie Charcot i Pierre Marie a París, França i Howard Henry Tooth a Cambridge, Anglaterra. La malaltia de CMT, també coneguda com a neuropatia hereditària motriu i sensitiva o atròfia de peroneus, abasta un grup de trastorns que afecten els nervis perifèrics. Els nervis perifèrics resideixen fora del cervell i la medul·la espinal i proveeixen informació als músculs i els òrgans sensorials de les extremitats. Els trastorns que afecten els nervis perifèrics es diuen neuropaties perifèriques.[1]
Símptomes
[modifica]La neuropatia de la malaltia de CMT afecta els nervis motors i sensorials. Una característica típica inclou debilitacions en els peus i dels músculs inferiors de la cama, que poden donar lloc a una deformació del peu i generar una marxa a passos grans que desencadena en ensopegades o caigudes freqüents. Les deformitats del peu, tals com arcs alts i dits en martell (una condició en la qual la conjuntura central d'un dit del peu es doblega cap amunt) són també característiques a causa de la debilitat dels músculs més petits del peu. A més, la part inferior de les cames pot adquirir un aspecte "d'ampolla de xampany invertida" a causa de la pèrdua de massa muscular. Conforme progressa la malaltia, poden ocórrer debilitats i atròfies musculars en les mans, donant com a resultat dificultats en les capacitats motores. Encara que els nervis sensitius també estan involucrats, molt rares vegades els pacients sofreixen d'entumiments o dolors significatius.
L'inici dels símptomes ocorre més sovint en l'adolescència o al començament de l'edat adulta. No obstant això, la seva incidència es pot retardar fins a mitjans de l'edat adulta. La gravetat dels símptomes és absolutament variable en diversos pacients i algunes persones mai arriben a adonar-se del trastorn. La progressió dels símptomes és molt gradual. La CMT no és letal i els pacients que pateixen dels tipus més comuns de CMT posseïxen una expectativa de vida normal.
Causes
[modifica]Una cèl·lula nerviosa comunica informació a diversos punts enviant senyals elèctrics a través d'una banda llarga i fina de la cèl·lula anomenada àxon. Per a augmentar la velocitat amb la qual viatgen aquests senyals elèctrics, l'àxon està recobert de mielina, que és produïda per altre tipus de cèl·lula anomenada cèl·lula de Schwann. La mielina s'embulla al voltant del axón com una espècie de rosca de gelea prevenint així la dissipació dels senyals elèctrics. Sense un axón i una capa de mielina intactes, les cèl·lules nervioses perifèriques no poden activar els músculs o retransmetre informació sensorial de les extremitats al cervell.
La malaltia de CMT és causada per mutacions en els gens que produïxen les proteïnes relacionades amb l'estructura i la funció bé sigui de l'àxon del nervi perifèric o de la capa de mielina. Encara que en diversos tipus de la malaltia de CMT diverses proteïnes són anormals, totes les mutacions afecten la funció normal dels nervis perifèrics. Per tant, aquests nervis es degeneren i perden lentament la capacitat de comunicar-se amb els diversos membres. La degeneració dels nervis motrius dona lloc a la debilitat del múscul i a atròfies en les extremitats (braços, cames, mans o peus), i la degeneració dels nervis sensitius comporta a una reducció en les sensacions de calor, fred i dolor.
Les mutacions genètiques en la malaltia de CMT són generalment hereditàries. Unes de tipus autosòmica dominant, d'altres de forma autosòmica recessiva, i altres per herència lligada al cromosoma X, En rares ocasions, la mutació genètica que causa la malaltia de CMT és una nova mutació que ocorre espontàniament en el material genètic del pacient i no ha estat transmesa hereditàriament.
Tipus
[modifica]Hi ha moltes formes de la malaltia de CMT. Els tipus principals inclouen CMT1, CMT2, CMT3, CMT4, i CMTX.
CMT1
[modifica]CMT1 és el tipus més freqüent i resulta d'anormalitats en la capa de mielina. Hi ha tres tipus principals de CMT1. CMT1A és una malaltia autosòmica dominant que resulta d'una duplicació del gen en el cromosoma 17 que posseïx instruccions per a produir la proteïna 22 de la mielina (PMP-22). La proteïna PMP-22 és un component essencial de la capa de mielina. Un excés d'aquest gen fa que l'estructura i la funció de la capa de mielina siguin anormals. Els pacients pateixen de debilitat i atròfia dels músculs inferiors de les cames a partir de l'adolescència; i més endavant pateixen de debilitats en les mans i la pèrdua de sensació. Cap destacar que existeix una neuropatia diferent de la neuropatia hereditària CMT1A coneguda com a neuropatia hereditària amb predisposició a paràlisi compressiva (HNPP, per les seves sigles en anglès) que és causada per l'absència d'un dels gens PMP-22. En aquest cas, la presència de nivells anormalment baixos del gen PMP-22 dona com a resultat neuropaties desmielinitzants episòdiques i recurrents. La CMT1B és una malaltia autosòmica dominant causada per mutacions dels gens que posseïxen instruccions per a produir la proteïna zero (P0) de mielina, que és altre component crític de la capa de mielina. La majoria d'aquestes mutacions són mutacions puntuals, el que significa que un error ocorre solament en una lletra del codi genètic de l'ADN. Fins a la data, els científics han identificat més de 30 mutacions puntuals diverses del gen P0. Com a resultat d'anormalitats en P0, la CMT1B produïx símptomes similars als de la CMT1A. El defecte del gen que causa la CMT1C, que també produïx símptomes similars als de la CMT1A, encara no ha estat identificat.
CMT2
[modifica]CMT2 és menys comuna que CMT1 i sorgeix d'anormalitats en l'àxon de la cèl·lula nerviosa perifèrica en lloc d'en la capa de mielina. Es va identificar recentment una mutació en el gen que codifica la proteïna 1B-beta del membre de la família de cinesines en casos de CMT2A. Les cinesines són les proteïnes que actuen com motors per a ajudar a accionar la transmissió de materials a través dels microtúbuls de la cèl·lula. Recentment es va identificar una altra mutació en el gen del neurofilament en una família russa amb CMT2I. Els neurofilaments són les proteïnes estructurals que ajuden a mantenir la forma normal de la cèl·lula. Els gens que causen altres tipus de CMT2 encara no s'han identificat.
CMT3
[modifica]La malaltia de CMT3 o de Dejerine-Sottas és una neuropatia desmielinitzant greu que comença en la infància. Els bebés pateixen d'atròfies i debilitats musculars severes i problemes sensorials. Aquest trastorn poc comú pot ésser a causa d'una mutació específica puntual del gen P0 o a una mutació puntual del gen PMP-22.
CMT4
[modifica]CMT4 abasta diversos subtipus diversos de neuropaties desmielinitzants autosòmiques recessives motores i sensorials. Cada subtipus de la neuropatia és causat per una mutació genètica diferent, pot afectar una població ètnica particular i produïx característiques fisiològiques o clíniques distintes. Els pacients que pateixen de CMT4 generalment presenten símptomes de debilitat en les cames durant la infantesa i poden perdre la capacitat de caminar en l'adolescència. Les anormalitats genètiques responsables de CMT4 encara no han estat identificades.
CMTX
[modifica]CMTX és una malaltia dominant relacionada al cromosoma X i és causada per una mutació puntual en el gen de la connexina 32 al cromosoma X. La proteïna connexina 32 es presenta en les cèl·lules de Schwann, les quals recobrixen els àxons del nervi, constituint un sol segment de la capa de mielina. Aquesta proteïna es pot relacionar a la comunicació de la cèl·lula de Schwann amb l'axó. Els barons que hereten un gen transformat de les seves mares presenten símptomes moderats a greus de la malaltia que comença al final de la infantesa o en l'adolescència (el cromosoma Y que els barons hereten dels seus pares no té el gen connexina 32). Les nenes que hereten un gen transformat d'un pare i un gen normal de l'altre pare poden desenvolupar símptomes lleus en l'adolescència o més tard, o no desenvolupar cap símptoma de la malaltia.
Diagnòstic
[modifica]El diagnòstic de la malaltia de CMT comença amb una història estàndard del pacient, antecedents familiars, i un examen neurològic. Se li pregunta al pacient sobre la naturalesa i durada dels seus símptomes i si altres membres de la família pateixen la malaltia. Durant l'examen neurològic un metge busca signes de debilitat muscular en els braços, cames, mans i peus, una disminució de la massa muscular, reflexos reduïts del tendó i pèrdua de sensibilitat. Els metges busquen signes de deformitats del peu, tals com arcs alts, dits en martell, taló invertit o peus plans. Altres problemes ortopèdics, tals com escoliosi o displàsia lleu del maluc, també poden presentar-se. Un símptoma específic que es pot trobar en pacients amb CMT1 és un engruiximent dels nervis que es pot palpar i fins i tot veure's a través de la pell. Aquests nervis engrandits, anomenats nervis hipertròfics, són causats per capes de mielina de gruix anormal.
Si se sospita CMT, el metge pot sol·licitar que es realitzin proves d'electrodiagnòstic en el pacient. Aquesta prova consta de dues parts: estudis de la conducció nerviosa i una electromiografia (EMG). En els estudis de la conducció nerviosa, es col·loquen elèctrodes en la pell sobre un nervi perifèric motor o sensitiu. Aquests elèctrodes produïxen una descàrrega elèctrica petita que pot causar un malestar lleu. Aquest impuls elèctric estimula els nervis sensitius i motors i proporciona informació quantificable que el metge pot utilitzar per a realitzar un diagnòstic. L'EMG involucra inserir un elèctrode tipus agulla a través de la pell per a amidar l'activitat bioelèctrica dels músculs. Les anormalitats específiques en les lectures indiquen una degeneració de l'àxon. L'EMG pot ser útil per identificar més de ple la distribució i gravetat de l'involucrament dels nervis perifèrics.
Si la resta de les proves semblen suggerir que un pacient té CMT, un neuròleg pot realitzar una biòpsia del nervi per a confirmar el diagnòstic. Una biòpsia del nervi implica llevar una part petita del nervi perifèric a través d'una incisió en la pell. Això es realitza amb major freqüència llevant una part del nervi situat al llarg del panxell. El nervi llavors s'examina en el microscopi. Típicament, els pacients amb CMT1 presenten signes de mielinització anormal. Específicament, poden presentar-se les formacions tipus "bulb de ceba", les quals posseïxen àxons recoberts per capes de cèl·lules de Schwann desmielinitzants i remielinitzants. Els pacients amb CMT2 mostren generalment una degeneració de l'àxon.
Es troben disponibles proves genètiques per a alguns tipus de CMT i molt aviat podrien estar disponibles per a detectar altres tipus; aquestes proves es poden utilitzar per a confirmar un diagnòstic. A més, està disponible un assessorament genètic per als pares que temen que puguin transmetre gens mutants als seus fills.
Tractament
[modifica]No existeix cura per a la malaltia CMT, però la teràpia física, teràpia ocupacional, fèrules i altres dispositius ortopèdics, i fins i tot la cirurgia ortopèdica, poden ajudar els pacients a enfrontar els símptomes incapacitants de la malaltia.
La teràpia física i ocupacional, el tractament preferit per a CMT, inclou l'exercici per a l'enfortiment muscular, estirar el múscul i els lligaments, proves d'estamina i exercici aeròbic moderat. La majoria dels terapeutes recomanen un programa de tractament especialitzat dissenyat amb l'aprovació del metge del pacient per a donar resposta a les capacitats i necessitats individuals. Els terapistes també suggereixen iniciar un programa de tractament precoç; l'enfortiment muscular pot retardar o reduir l'atròfia del múscul, pel que l'enfortiment muscular és més útil si es comença abans que la degeneració del nervi i l'augment en la debilitat del múscul acabin en incapacitació.
Els exercicis d'estirament poden prevenir o reduir les deformitats comunes que resulten d'una acció no uniforme del múscul sobre els ossos. Els exercicis que ajuden a augmentar l'estamina o la resistència muscular contribuïxen a prevenir la fatiga que resulta de realitzar activitats diàries que requereixen força i mobilitat. L'activitat aeròbica moderada pot ajudar a mantenir una bona condició cardiovascular i una bona salut en general. La majoria dels terapeutes recomanen exercicis de baix impacte o de zero impacte, tals com la bicicleta i la natació, en lloc d'activitats tals com caminar o trotar, que poden col·locar tensió en els músculs i en les conjuntures.
Molts pacients de CMT requereixen fèrules per al turmell i altres dispositius ortopèdics per a mantenir la mobilitat diària i prevenir lesions. Les fèrules del turmell poden ajudar a prevenir esquinços (torçades) proporcionant ajuda i estabilitat durant activitats tals com caminar o pujar escales. Els botins o les botes altes també poden ajudar els pacients amb turmells febles. Les fèrules del polze poden ajudar a combatre la debilitat de la mà i la pèrdua de capacitats motores fines. Els dispositius d'ajuda han de ser utilitzats abans que empitjori la incapacitació, ja que poden prevenir la tensió del múscul i reduir-ne la debilitació. Alguns pacients amb CMT poden optar per una cirurgia ortopèdica per a invertir deformitats del peu i les conjuntures.
Referències
[modifica]- ↑ Krajewski, K. M. «Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A». Brain, 123, 7, 2000, pàg. 1516–27. DOI: 10.1093/brain/123.7.1516. PMID: 10869062.
- National Institute of Neurological Disorders and Stroke Arxivat 2007-04-09 a Wayback Machine.: publicat sota domini públic.